Held in romantically beautiful Lisbon, this AD/PD was the biggest thus far, drawing 3,982 attendees from 73 countries, according to Abraham Fisher, its longtime lead organizer. But as the field grows, it has little to celebrate, and the mood reflected that. Trialists were appealing for patience and pointing to deepening clinical trials skills as they learn from continuing setbacks on investigational therapies, most recently of lanabecestat, crenezumab, and aducanumab. Trials targeting tau were not yet reading out, but the medical food Souvenaid posted modestly positive results. At the other end of the bench-to-bedside spectrum, basic science is diversifying. Next-gen genetics is spilling out gene variants which, together with RNAseq, are energizing research into glial and lipid biology. Debate about the amyloid hypothesis, a fixture at AD/PD, evolved toward a focus on genetic variability in how a person’s innate immune system responds to amyloid deposition. Aβ itself? Nothing special about it. It aggregates into an irritant as the brain’s ability to degrade it wanes with age. It starts things off, but other factors later dominate the disease. Or so researchers now think. Read Madolyn Rogers and Gabrielle Strobel’s coverage.
At AD/PD Conference, New Alzheimer’s Genes Reinforce Known Pathways
Genome-wide association studies have turned up some 30 loci linked to Alzheimer’s, yet GWAS still left much of the disease’s heritability unexplained. To find the remaining genes, geneticists have turned to whole-genome and whole-exome sequencing, to family cohorts, and to studies of cell-type specific gene expression. After years of grunt work assembling cohorts and finessing techniques, those approaches are finally bearing fruit, and at the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Portugal’s beautiful capital city of Lisbon, the fruit were on display. Researchers unveiled a trove of new genetic associations, as well as some mechanistic explorations (see Part 2 of this story).
“We are learning to navigate the post-GWAS world,” Alexandre Amlie-Wolf of the University of Pennsylvania, Philadelphia, summed it up.
Taken together, the new genetic associations emphasize the primacy of APP processing and innate immunity for AD pathogenesis. In fact, talks and posters throughout this five-day conference suggested overall that the way a person’s microglia respond to amyloidosis may determine whether they develop Alzheimer’s disease. Microglia may influence Parkinson’s disease as well, with some talks in Lisbon linking these brain-resident immune cells to PD-risk-gene expression.
Navigating the Pre-GWAS World. Down the road from the AD/PD conference center, in Belém’s Jeronimos Monastery, lies Vasco Da Gama, who navigated the 15th-century seas to discover the first ocean route from Europe to India, among other feats. Detail from his carved tomb.
Taking a bird’s-eye view to the known genetic associations, it looks as if age-related neurodegenerative disease in general arises due to a failure to respond effectively to cellular damage, said John Hardy of University College London. When a cellular clearance mechanism starts to slow down, the most abundant protein normally cleared in that way—Aβ in AD, α-synuclein in PD, tau in primary tauopathies—will accumulate and fall out of solution, forming aggregates. “Perhaps there’s nothing special about these proteins,” Hardy said. In other words, their specific cellular functions may have little to do with disease pathogenesis. This conclusion is indeed what years of research into their cellular and molecular biology amount to, to the extent that their functions are even understood. Perhaps researchers should instead focus on boosting their clearance to ameliorate disease, Hardy suggested.
The New Gold Standard—Whole-Exome and -Genome Sequencing
The go-to tool for geneticists nowadays is to sequence whole genomes or exomes from large cohorts of people. They often select groups at high risk of disease, such as families with a history of dementia. Unlike GWAS, which find common variants that each contribute little risk, next-generation sequencing can turn up rare variants that confer high risk. The Alzheimer’s Disease Sequencing Project recently identified two new risk genes from whole-exome analysis of 11,000 cases and controls (Aug 2018 news). Although such genes contribute little to population risk, they can illuminate disease pathways. Besides uncovering new genes, whole-exome sequencing finds rare variants in AD’s canonical autosomal-dominant genes that confer less disease risk, for example presenilin 1 mutations associated with late-onset disease. “Whole-exome and -genome sequencing have changed genetic research,” said Christine van Broeckhoven of the University of Antwerp, Belgium.
In Lisbon, Lindsay Farrer of Boston University presented the latest findings from the ADSP. To increase their odds of finding rare genes, the researchers selected “enriched” cases, i.e., people with Alzheimer’s who also had at least one close relative with the disease. They compared whole-exome data from 679 such cases and 5,094 controls. The most robust finding was a single rare missense variant in caspase-7 (CASP7) that associated with AD. For other genes, individual variants missed statistical significance, but the occurrence of several different coding variants linked with disease flagged the locus as a likely risk factor. The genes ANXA5, AARD, IGHJ6, and C1orf173 all passed this “gene-based” association test for AD risk (Zhang et al., 2019).
Plethora of New AD Genes. Whole-exome sequencing has identified 24 genes with AD-linked coding variants; asterisks indicate the 18 found by ADSP. [Courtesy of Lindsay Farrer.]
What do these genes do? ANXA5 is part of endocytosis and IGHJ6 of immunity, while the functions of AARD and C1orf173 are unknown. CASP7 has been implicated in AD genetics before (Shang et al., 2015). This caspase might contribute to disease by more than one mechanism. Farrer noted that it snips amyloid precursor protein to create a toxic C31 fragment (Fiorelli et al., 2013). In addition, CASP7 helps activate microglia, again underscoring the importance of these cells in AD (Burguillos et al., 2011; Ayers et al., 2016).
In another gene-hunting tack reported at AD/PD, ADSP researchers searched whole exomes for rare variants predicted to harm protein function that occurred only among the 5,617 cases, never in the 4,594 controls. This approach unearths high-penetrance variants that are often lost in traditional genetic studies, Farrer explained. The most frequent variant found in this way occurred in 10 AD patients. It is a NOTCH3 missense mutation predicted to strengthen notch3 binding to its ligand, Jagged-1. NOTCH3 signaling is regulated by γ-secretase and BACE1; a common synonymous mutation in the gene was linked to late-onset Alzheimer’s before (Sassi et al., 2018).
In addition, four participants with AD carried the TREM2 variant Q33X, which causes the bone disorder Nasu-Hakola disease when homozygous. All had late-onset AD and no sign of bone cysts. The researchers also found 10 people with AD who inherited an ATP-binding cassette sub-family D member 4 (ABCD4) haplotype containing three rare variants, and another eight people who inherited multiple variants in both cadherin EGF LAG Seven-pass G-type receptor 1 (CELSR1) and the nearby G2 and S phase-expressed protein 1 (GTSE1). The laminin subunit γ3 (LAMC3) and titin (TTN) genes also sported an unusual number of deleterious variants in AD cases (Patel et al., 2019).
This is but a list of genes—does their biology support an association with AD? ABCD4 mutations cause vitamin B12 levels to fall, and B12 deficiency is a risk factor for the disease, as are high levels of its substrate, homocysteine (Mar 2002 news; Grarup et al., 2013; Chen et al., 2015). CELSR1 plays a role in brain development and neural tube defects, while GTSE1 regulates microtubule stability. Laminin distribution is perturbed in AD brain, and variants have been linked to onset age (Palu and Liesi, 2002; Saad et al., 2015). Titin, a muscle protein also known as connectin, appears to have no link to AD, but has been shown to form amyloids in vitro (Marsagishvili et al., 2005; Bobylev et al., 2016).
Altogether, ADSP whole-exome sequencing has added 18 new AD risk genes to the catalogue so far, Farrer said (see image above). Newly found as they may be, they all the same fall into the familiar functional categories of innate immunity, APP processing, vesicle trafficking, and neuronal signaling. “We are filling in genes in well-established pathways that lead to Alzheimer’s disease,” Farrer concluded.
Mining Family Data
By sequencing DNA from families with a disproportionate AD burden, geneticists can also boost their odds of finding rare variants, even in smaller cohorts. In Lisbon, Margaret Pericak-Vance of the University of Miami, Florida, described the ADSP’s Discovery data set, which comprises whole-genome data from 67 such Caribbean Hispanic and 46 Caucasian families. In the former, Pericak-Vance found an A kinase anchor protein 9 (AKAP9) variant that tracked with disease in two families, as well as missense variants in myelin regulatory factor (MYRF) and asparaginase-like 1 (ASRGL1) (Vardarajan et al., 2018). The latter group harbored putatively deleterious variants in nitric oxide synthase 1 adaptor protein (NOS1AP), ATP binding cassette transporter 1 (ABCA1), FISP2, and the long noncoding RNA RP11-433J8 (Beecham et al., 2018). In both ethnic groups, new variants in known AD genes cropped up, as well.
AKAP9 stabilizes microtubules and was previously linked to AD in African American families (Aug 2013 conference news; Logue et al., 2014). ABCA1 is a known AD gene that regulates cholesterol efflux and ApoE levels (Aug 2004 news; Oct 2005 news). The other genes were not previously associated with AD.
Also at AD/PD, Richard Mayeux of Columbia University, New York City, presented two other genes. Whole-exome sequencing of 31 Caribbean Hispanic families nabbed 10 different rare mutations in Sfn2-related CREBBP activator protein (SRCAP) (Vardarajan et al., 2017). Mutations in SRCAP cause Floating-Harbor syndrome, a rare condition marked by stunted growth, and SRCAP protein has been linked to ALS (May 2013 news). SRCAP activates CREB binding protein and helps repair DNA. The second gene, ceroid lipofuscinosis 5 (CLN5), has a missense variant that segregates with AD in these families and causes problems with retromer trafficking (Qureshi et al., 2018).
In ongoing work, the ADSP is mining a larger whole-genome data set called Discovery Extension, which so far includes 70 Caribbean Hispanic, 58 Caucasian, and 10 African American families, Pericak-Vance said. The team is adding in the genomes from 34 African American families participating in UMiami’s Research in African American Alzheimer’s Disease Initiative as well as 26 families from the Puerto Rican Alzheimer’s Disease Initiative (PRADI). The combined data set has turned up a linkage region on chromosome 5 in African American and Dutch families, as well as a disease link on chromosome 12 near LRRK2 in one African American family. In Puerto Rican families, a region on chromosome 9 near C9ORF72 segregates with disease, but it lacks the infamous repeat expansion. In each case, the genes responsible for the disease association have yet to be identified. The ADSP plans to expand this analysis to nearly 1,000 families, Pericak-Vance said.
In a similar vein, Julie Hoogmartens of the University of Antwerp used whole-genome sequencing of 19 people with early onset AD to search for new recessive familial genes. Working with van Broeckhoven, Hoogmartens found 113 rare homozygous coding variants in them, then narrowed the list to 13 that replicated in an additional 353 cases but did not appear in 903 controls. She winnowed down further to those that were expressed in brain and predicted to be deleterious. This produced four candidate genes: C1ORF194, CCDC136, GFAP, and VWA2, aka Von Willebrand Factor A Domain-Containing Protein 2.
Hoogmartens is particularly interested in VWA2. Found in exosomes, this extracellular matrix protein is considered part of the innate immune response (Dahmer et al., 2016). She verified its Alzheimer’s association in a Belgian cohort of 1,253 and a European cohort of 814 AD patients. Five people carried a homozygous VWA2 variant. Five others carried two different deleterious variants, one from their mother and one from their father, thus damaging both of their VWA2 alleles. This reflects a recessive mode of inheritance, Hoogmartens said. Next, she will examine how the variants affect gene expression, and compare VWA2 protein levels in AD patients and controls.
Tracking Down Elusive Protective Genes
Some populations are known to resist the effects of AD risk genes, and family data can help scientists discover the protective genes at play. In Lisbon, Christiane Reitz of Columbia University noted that even though Caribbean Hispanics run twice the risk of AD as Caucasians, the ApoE4 allele has a less harmful effect on them (Tang et al., 2001; Olarte et al., 2006). To search for modifying factors, Reitz’s colleague Badri Vardarajan examined whole-genome sequence data from healthy elderly ApoE4 homozygotes in two Caribbean Hispanic cohorts. These were WHICAP, a longitudinal community study in New York City, half of whose 6,000 participants are Hispanic, and the Estudio Familiar de Influencia Genética en Alzheimer (EFIGA) cohort of 500 multiplex families from the Dominican Republic, Puerto Rico, and New York City. Both studies follow participants at regular intervals. In 70 homozygous and 130 heterozygous E4 carriers from EFIGA, Vardarajan and Reitz looked for coding variants that appeared in at least 5 percent of cognitively healthy E4 homozygotes but not in people with AD. They verified in the larger WHICAP study that these variants were either absent or less frequent in symptomatic people. This produced 23 variants, which researchers examined in 173 independent AD families and 500 sporadic AD cases for correlations with either AD risk or age of onset. They came up with two that tracked with onset: BMP1 and NBEAL1.
NBEAL1 had the largest effect, delaying symptoms. The neurobeachin-like 1 protein is involved in vesicle trafficking and receptor signaling and correlates with white-matter hyperintensities (Jian et al., 2018; Traylor et al., 2016). The gene resides near an age-of-onset ADGC GWAS hit (Naj et al., 2014). Reitz said the groups are investigating what NBEAL1 does.
Jeffery Vance, University of Miami, is also pursuing genes that protect against ApoE4; however, he is using a different strategy and an additional ethnic group. Like Hispanics, African Americans seem partially resistant to the deleterious effects of an E4 allele (Farrer et al., 1997; Evans et al., 2003; Weuve et al., 2018). To track down the gene(s) responsible, Vance exploits the fact that African American and Hispanic gene pools began to mix only about 300 years ago, owing to colonialism and slavery. In the U.S., Hispanic populations typically carry a mixture of European, African, and Native American genes, with the former predominating, while African Americans carry genes from several African tribes as well as some European DNA (Zakharia et al., 2009). Different modern groups carry distinct proportions. For example, Haitians tend to have mostly African ancestry, while Puerto Ricans have mostly European.
This allowed Vance to ask if protective associations were the result of local ancestry, i.e., a variant that lies near ApoE and thus is inherited with it, or global ancestry, i.e., some separate factor that was widespread among a particular group because of the part of the world in which they arose. The researchers analyzed 1,766 cases and 3,730 controls of African American ancestry, and 220 cases and 169 controls of Puerto Rican ancestry. For both ethnic groups, the analysis strongly suggested that local, not global, ancestry was modifying ApoE risk. In other words, ApoE4 alleles on an African local background conferred less risk than ApoE4 alleles on a European local background, no matter which ethnic group a person belonged to (Rajabli et al., 2018).
Vance is now trying to track down the protective variant by identifying sequence differences in this local ancestry region and correlating them with functional changes in gene expression or pathology, using postmortem brain samples from homozygous ApoE4 carriers from each population. The researchers have found more than 700 sequence differences in the 2 megabase region around ApoE, all of them noncoding. Because the TOMM40 gene occurs near ApoE and has been linked to differences between African and European AD risk before, Vance investigated its effect (Roses et al., 2014; Yu et al., 2017). However, in Lisbon he said that he found no relationship between TOMM40 alleles and AD risk in African Americans, although among people with European local ancestry and an ApoE3 allele, the very long poly-T repeat of TOMM40 did correlate with lower risk. In answer to audience questions, Vance said he still has not identified the variant that protects against ApoE4, but he believes it will turn out to be a regulatory change in ApoE expression.—Madolyn Bowman Rogers
Grarup N, Sulem P, Sandholt CH, Thorleifsson G, Ahluwalia TS, Steinthorsdottir V, Bjarnason H, Gudbjartsson DF, Magnusson OT, Sparsø T, Albrechtsen A, Kong A, Masson G, Tian G, Cao H, Nie C, Kristiansen K, Husemoen LL, Thuesen B, Li Y, Nielsen R, Linneberg A, Olafsson I, Eyjolfsson GI, Jørgensen T, Wang J, Hansen T, Thorsteinsdottir U, Stefánsson K, Pedersen O.
Genetic architecture of vitamin B12 and folate levels uncovered applying deeply sequenced large datasets.
PLoS Genet. 2013 Jun;9(6):e1003530. Epub 2013 Jun 6
PubMed.
Bobylev AG, Galzitskaya OV, Fadeev RS, Bobyleva LG, Yurshenas DA, Molochkov NV, Dovidchenko NV, Selivanova OM, Penkov NV, Podlubnaya ZA, Vikhlyantsev IM.
Smooth muscle titin forms in vitro amyloid aggregates.
Biosci Rep. 2016 Jul;36(3) Print 2016 Jul
PubMed.
Vardarajan BN, Barral S, Jaworski J, Beecham GW, Blue E, Tosto G, Reyes-Dumeyer D, Medrano M, Lantigua R, Naj A, Thornton T, DeStefano A, Martin E, Wang LS, Brown L, Bush W, van Duijn C, Goate A, Farrer L, Haines JL, Boerwinkle E, Schellenberg G, Wijsman E, Pericak-Vance MA, Mayeux R, Alzheimer's Disease Sequencing Project, Wang LS.
Whole genome sequencing of Caribbean Hispanic families with late-onset Alzheimer's disease.
Ann Clin Transl Neurol. 2018 Apr;5(4):406-417. Epub 2018 Mar 13
PubMed.
Beecham GW, Vardarajan B, Blue E, Bush W, Jaworski J, Barral S, DeStefano A, Hamilton-Nelson K, Kunkle B, Martin ER, Naj A, Rajabli F, Reitz C, Thornton T, van Duijn C, Goate A, Seshadri S, Farrer LA, Boerwinkle E, Schellenberg G, Haines JL, Wijsman E, Mayeux R, Pericak-Vance MA, Alzheimer's Disease Sequencing Project.
Rare genetic variation implicated in non-Hispanic white families with Alzheimer disease.
Neurol Genet. 2018 Dec;4(6):e286. Epub 2018 Nov 21
PubMed.
Logue MW, Schu M, Vardarajan BN, Farrell J, Bennett DA, Buxbaum JD, Byrd GS, Ertekin-Taner N, Evans D, Foroud T, Goate A, Graff-Radford NR, Kamboh MI, Kukull WA, Manly JJ, Alzheimer Disease Genetics Consortium, Alzheimer Disease Genetics Consortium.
Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans.
Alzheimers Dement. 2014 Nov;10(6):609-618.e11. Epub 2014 Aug 27
PubMed.
Expression, Expression, Expression—Time to Get on Board with eQTLs
Next-generation genetic analysis has opened the floodgates to a wave of new gene discovery. Whole-exome and -genome sequencing have nearly doubled the number of previously known Alzheimer’s genes (see Part 1 of this story). But modern geneticists have other tools available as well. One is to look for changes in gene expression in distinct brain cell populations to understand how risk variants might make people sick. “We are moving from generating genomic data to transcriptomic data,” said Elisa Navarro of the Icahn School of Medicine at Mount Sinai, New York.
This approach is important, because many disease associations fall into regulatory, not coding regions. When variants affect gene expression, geneticists call them expression quantitative trait loci, and eQTLs have become a big deal in AD genetics. To find eQTLs, geneticists correlate variants with expression changes in a given cell type. eQTLs are cell-type-specific because cell types tend to contain a unique mix of different transcription factors that bind distinct enhancers. Thus, a mutation in a particular enhancer may only affect expression in one type of cell, and eQTLs therefore offer clues to the cell type most involved in the disease. For example, for Alzheimer’s disease, researchers have now linked much of the genetic risk to eQTLs in microglia (Jun 2017 news).
At the Heart of it All. Surprisingly many AD gene variants exert their effect in microglia, where they influence the response to amyloidosis. [Courtesy of Annerieke Sierksma.]
In Lisbon, speakers presented similar findings for Parkinson’s disease. Navarro, working with Towfique Raj at Icahn, used monocytes as a more accessible proxy for microglia. She isolated monocytes from 60 people with PD and 57 aged-matched controls, and analyzed their gene expression with RNAseq. Monocytes from patients showed numerous expression changes relative to control cells, confirming dysregulation of the immune system in this disease.
Pathway analysis flagged lysosomal and mitochondrial genes as the most altered. Most of the lysosomal genes were downregulated, and indeed, cultured monocytes from patients degraded waste only sluggishly. Notably, both these pathways contain many GWAS hits for PD. So far, the researchers have identified, in these monocytes, three eQTLs that affect expression of a known PD gene—LRRK2, GPR65, and GPNMB (Ramdhani et al., 2018). Navarro’s study is underpowered to find eQTLs, but the researchers are adding samples from another 120 people, Navarro said. Though the findings implicate immune cells in PD pathogenesis, they provide no information on how gene expression may change in the brains of people with the disease.
Regina Reynolds, working with Mina Ryten of University College London, took a different dig at the biology underlying GWAS associations. She examined whether genes linked to PD were expressed more or less highly in different brain cell types—neurons, astrocytes, oligodendrocytes, or microglia. She saw no consistent difference. Then the researchers dug for any association of PD genes with chromatin regulation in different cell types, and again came up empty. “We were getting a bit frustrated,” Reynolds said in Lisbon.
The researchers reframed the question. Perhaps instead of Parkinson’s risk being concentrated in a particular cell type, it is in particular processes that are ubiquitous to all cells, such as autophagy, lysosomal degradation, or mitochondrial function. With that, they hit pay dirt, when pathway analysis placed Parkinson’s heritability predominantly in lysosomal genes. But do lysosomes function the same in every cell type? Here the analysis turned up a difference, finding higher expression of lysosomal genes in microglia than in neurons (Reynolds et al., 2018). “We should be using glial cells in our Parkinson’s models,” Reynolds concluded.
For Alzheimer’s disease, too, evidence is converging on microglia as a key factor (see image above). In Lisbon, Annerieke Sierksma and Ashley Lu of KU Leuven, Belgium, presented their analysis of how AD risk genes change expression in the presence of amyloid or tau pathology. Working with Bart De Strooper and Mark Fiers, the researchers compared gene expression in APPswe/PS1L166P and THY-Tau22 mice. In the latter, only a handful of AD genes changed expression. These were mostly involved in neuronal function; they were suppressed, and were suppressed stably over the life of the mouse.
By contrast, in the amyloid mice, the researchers found a massive increase in AD gene expression as amyloidosis advanced. Most of these genes clustered in the inflammatory pathway and were expressed in microglia. In addition to known genes, the analysis highlighted 11 new genes of interest: GPC2, TREML2, SYK, GRN, SLC2A5, SAMSN1, PYDC1, HEXB, RRBP1, LYN, and BLNK. All are expressed in microglia, controlled by the master regulator PU.1. All rev up in the presence of amyloid, but have not been previously linked to AD. “AD risk variants determine how microglia respond to accumulating Aβ pathology,” the researchers concluded. The data underline the idea that amyloid triggers Alzheimer’s disease (Sierksma et al., 2019).
Other researchers agreed, saying privately that their own ongoing human-expression studies indicate extraordinarily broad gene-expression increases in response to amyloid deposition, and much more limited expression changes in response to tau pathology.
John Hardy of University College London agrees with this overall conclusion. “Genetic variability in the response to amyloid is the key to AD,” he told Alzforum. Hardy noted that many AD genes affect the metabolism of membrane lipids and motility of microglia. Perhaps amyloidosis starts in the cell membrane, and cells then have to respond to membrane disruption, he proposed.
In Parkinson’s disease, on the other hand, lysosomal stress seems to be the key. Genetic variants that boost α-synuclein expression are enough to overload the lysosome (Nov 2003 news; Jun 2010 news). In tauopathies, such as frontotemporal dementia and progressive supranuclear palsy, the ubiquitin proteasome is most affected. This organelle bears primary responsibility for mopping up tau, and again, high levels of tau alone are enough to trigger pathology (Rovelet-Lecrux et al., 2010). “We’ve come to the same conclusion in all three diseases: an age-dependent failure of clearance pathways,” Hardy said.
Geneticists hope these insights will lead to new therapies. “Genetics can drive discovery of drug targets,” said Gerard Schellenberg of the University of Pennsylvania. “There is a lot of biology yet to be revealed.”—Madolyn Bowman Rogers
APP Upp: Mutation Nixes Six Amino Acids from Aβ, Spurs Aggregation
Thirty missense mutations in amyloid precursor protein are known to cause autosomal-dominant Alzheimer’s disease. Now, scientists at Uppsala University, Sweden, have identified a deletion in the APP gene that does the same thing. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, María Pagnon de la Vega described a mutation that leads to the loss of six amino acids from the mid-region of the Aβ peptide. It segregated with disease in a single family.
Cleavage of this mutant APP by BACE1 and then γ-secretase produces truncated peptides, Aβ34 and Aβ36. Intriguingly, the shortened Aβ36 peptide appears particularly prone to aggregation. “To our knowledge, this is the first deletion in APP found to cause autosomal-dominant AD,” Pagnon de la Vega said in Lisbon.
Working with Lars Lannfelt, Dag Sehlin, and Martin Ingelsson at Uppsala, Pagnon de la Vega identified the “Uppsala” deletion in a family whose affected members developed symptomatic AD at about 40 years of age. The researchers traced its inheritance through three generations, where the disease followed an autosomal-dominant pattern. Their study focused on two afflicted brothers, age 43 and 45, one of whom later died. The brothers had an affected parent who died of AD. They had typical tau tangles by neuropathology and elevated cerebrospinal fluid tau markers, as well as the expected brain atrophy and hypometabolism in temporal and parietal cortex.
Notably, however, their CSF Aβ, as measured by standard assays, resembled that of healthy controls. Ingelsson told Alzforum that the antibodies used in CSF assays also detect Aβ34 and Aβ36, as their epitopes are not in the deleted region. In contrast, PiB PET scans were mildly positive, with an SUVR of about 1.4. In the brother who came to autopsy, his cortical amyloid plaques contained mostly Aβ36 or smaller fragments and looked unusually round, with sharply defined edges. The researchers found almost no Aβ40 or Aβ42 in these deposits. They also found plaques in the occipital cortex, where few tend to form in most cases of sporadic AD.
When the researchers examined the behavior of synthetic Aβ34 and Aβ36 in vitro, they found that Aβ36 fibrillizes rapidly, far faster than wild-type Aβ42 or even Aβ containing the Arctic mutation.
Pagnon de la Vega noted that the pattern of aggregation is different as well. The researchers were unable to detect protofibrils, the intermediate stage of aggregation, by ELISA and electron microscopy; instead, fibrils popped up right away. The Swedish researchers have made a double transgenic UppSwe mouse; it accumulates mostly Aβ36 as well. The researchers are currently making antibodies against the shortened peptides.
To further characterize the new mutation, Pagnon de la Vega and colleagues transfected cell lines with Uppsala APP. They found high levels of Aβ34 in the medium, but no sign of the sAPPα cleavage fragment by Western blot. The deletion may shift α-secretase cleavage toward the N-terminus, so that the resulting fragment is no longer recognized by standard antibodies, Pagnon de la Vega surmised. She is currently analyzing the media by mass spectrometry to find out if α-secretase cleavage occurs.
Deletion, Where Art Thou? A newly discovered APP mutation leads to Aβ peptides that are missing six amino acids. Researchers at AD/PD were coy about which ones they are.
The audience in Lisbon was much intrigued by the findings, and expressed some dismay when the scientists would not disclose the location of the deletion.
The same group, led by Lannfelt, has identified previous APP mutations. The Swedish mutation alters two amino acids next to the BACE1 cleavage site and stimulates this cut, boosting production of Aβ40 and Aβ42 (Mullan et al., 1992). The Arctic mutation results in Aβ that readily forms protofibrils. People with this mutation develop AD early, but have few amyloid cored plaques and are negative by PiB PET. Arctic protofibrils were used to develop the BAN2401 antibody that has entered Phase 3 trials.
At the same location as the Arctic mutation, a single amino acid deletion has been linked to AD in three Japanese families (Tomiyama et al., 2008). This Osaka mutation appears recessive, and homozygous carriers have an unusual form of AD, with few plaques by PiB PET, and often develop motor problems and difficulty walking. This mutant Aβ readily forms oligomers, but no fibrils.
APP also comes in at least one protective variant; carriers of the Icelandic mutation produce about 20 percent less Aβ over their lifespan.—Madolyn Bowman Rogers
Parsing How Alzheimer’s Genetic Risk Works Through Microglia
Much of the genetic risk of Alzheimer’s disease plays out in microglia. But exactly how do risk variants change these cells? At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, speakers filled in some gaps, and discussion of this question buzzed in the hallways all week. Christian Haass of the German Center for Neurodegenerative Diseases in Munich characterized progranulin as TREM2’s opposite. While mutations in TREM2 suppress microglial activation, mutations in progranulin permanently rev them up into voracious, toxin-spewing machines. Other speakers associated GWAS hits in general, and the MS4A gene cluster in particular, with hyperactive, damaging microglia. Researchers also highlighted potential therapeutic targets among microglial genes. Two talks presented evidence that suppressing the CD33 receptor can contain amyloidosis (see Part 5 in this series), while another fingered BIN1 as a key culprit in propagating phosphorylated tau.
Modulating microglia for therapeutic purposes will be complicated, because these cells are exquisitely sensitive to their environment and can assume numerous different activation states depending on the stimuli around them. For example, Beth Stevens of Boston Children’s Hospital recently reported that different types of injury, such as demyelinating lesions and amyloid plaques, induce distinct microglial phenotypes (Dec 2018 news). In a plenary talk in Lisbon, Stevens noted, “There are disease-specific signatures. We need to be able to target these microglial subpopulations to switch them from a detrimental to a healthy state.”
GWAS Hits in Microglial Activation: An Up-Down Affair
Haass’ data suggests that interventions will need to be finely tuned. His lab previously reported that TREM2 normally activates microglia to respond to injury, and disease-linked variants disrupt this function, locking microglia into a homeostatic, unresponsive state (May 2017 news). In Lisbon, Haass showed that the opposite extreme is equally harmful. Julia Götzl, Matthias Brendel, and Georg Werner from his group, along with Anja Capell at Ludwig-Maximilians University in Munich, analyzed gene expression in microglia from progranulin knockout mice. The cells’ profile was diametrically opposed to that of TREM2 knockouts, with homeostatic genes suppressed and inflammatory genes up.
Polar Opposites. In aged TREM2 loss-of-function mice (left), microglia remain abnormally quiescent by TSPO scan, while in progranulin loss-of-function mice (right), they fire up. [Courtesy of Christian Haass.]
In keeping with this, microglia were more active sans progranulin. Progranulin-negative microglia gobbled up more bacteria than did wild-types in a dish, and migrated further in brain-slice cultures. This happened in mouse brain, as well. When APPPS1 mice were crossed with progranulin knockouts, many more microglia clustered around amyloid plaques in the progranulin-negative offspring. Both humans and mice with progranulin mutations had more hyperactivated microglia in their cortices, as seen by TSPO PET.
Importantly, however, these overly aggressive microglia were just as damaging to diseased brains as were the abnormally quiescent TREM2 knockouts. In essence, they mark the tips of a spectrum going from too little microglial response to amyloidosis to too much. Both types of knockout had weak brain glucose metabolism (Götzl et al., 2019, in press). The data suggest a narrow therapeutic window for tweaking microglia, where pushing too far in either direction could cause harm, Haass noted. Progranulin mutations cause frontotemporal dementia, not AD, although Haass’ group found that the protein rises in the cerebrospinal fluid of AD patients as disease progresses (Nov 2018 news).
Progranulin and TREM2 are far from the only genes associated with damaging microglial states. Nicola Fattorelli, working with Carlo Sala Frigerio, Leen Wolfs, and Bart De Strooper at the University of Leuven, Belgium, performed RNA-Seq on more than 10,000 single microglia isolated from the cortices and hippocampi of young and old APP NL-G-F knock-in and wild-type control mice. In Lisbon, Fattorelli showed that the gene-expression profiles defined three distinct types of microglia: homeostatic cells, which were most abundant in young mice; interferon-response microglia, which increased with age equally in wild-type and AD mice; and amyloid-response microglia (ARM).
Separate Fates. Gene-expression analysis of mouse microglia showed that homeostatic cells can transform into two distinct forms, one associated more with age (IRM), the other more with amyloidosis (ARM). [Courtesy of Cell Reports, Sala Frigerio et al.]
ARM were mostly absent in young wild-type mice, and increased to 10 percent of the microglial population with age. In young APP NL-G-F mice, already one-third of microglia were in the ARM state, and their numbers mushroomed to half by one year of age. This progression toward ARM happened faster in females than males.
Importantly, the expression profile of ARM cells was enriched for Alzheimer’s GWAS hits. ApoE and TREM2 were up, whereas BIN1, MS4A6B, CD33, PICALM, and INPP5D were down in these cells. The researchers found a similar expression profile in microglia from APP/PS1 mice, and also in the Accelerating Medicines Partnership-AD transcriptomic database of human brain samples. In the latter, ARM transcriptional changes correlated with plaque burden.
In mouse brain, immunostaining revealed that microglial ApoE expression mainly occurred around amyloid plaques, dropping off gradually in microglia that were farther away. To determine ApoE’s function here, the scientists examined aged APP/PS1 mice lacking ApoE. They found almost no ARM in them, suggesting that ApoE is required to induce this microglial state. The paper appears today in Cell Reports (Sala Frigerio et al., 2019).
Fattorelli told Alzforum that ARM resemble the disease-associated (DAM) or neurodegenerative phenotype (MGnD) microglia identified in other studies (Jun 2017 news; Sep 2017 news).
Haass noted that these findings fit with recent research from his group showing that microglia are the main source of ApoE in plaques (Jan 2019 news).
Meanwhile, Amanda McQuade, working with Mathew Blurton-Jones at the University of California, Irvine, implicated the microglial receptor MS4A6A in damaging activation states. How variants in the MS4A gene cluster act in neurodegeneration is unknown. Rather than mouse cells, McQuade used microglia generated from human iPSCs (Jul 2016 news; McQuade et al., 2018). When she knocked out MS4A6A in these cells, 28 out of 53 AD-linked genes significantly changed expression. These genes affect phagocytosis, chemotaxis, and immune response. In line with this, phagocytosis became sluggish.
Overall, the cells assumed a DAM-like gene expression profile, McQuade said. In ongoing work, she is transplanting these human cell lines into mouse brain to examine the consequences of MS4A6A knockout in vivo. In a new review, the scientists call for better microglial models that mimic the natural in vivo environment, in order to determine which microglial functions actually affect disease (McQuade and Blurton-Jones, 2019).
Where are the Drug Targets in All This?
MS4A may also affect disease through TREM2. Carlos Cruchaga previously reported that a noncoding variant, or eQTL (see Part 2 of this series), that boosts expression of MS4A4A and MS4A6A in blood and brain also raises levels of soluble TREM2 (sTREM2) in cerebrospinal fluid, while lowering AD risk and delaying age of onset (Jul 2018 news). In Lisbon, Cruchaga, of Washington University in St. Louis, added data supporting this relationship. In cell culture, he showed, overexpressing MS4A4A increased release of sTREM2, while knocking down MS4A4A suppressed it. This suggests that MS4A affects AD risk by modifying sTREM2 levels, Cruchaga believes. He noted that the strength of the association was comparable to ApoE’s effect on CSF Aβ42, and concluded that MS4A4A could offer a therapeutic handle.
Curiously, other data on the relationship between MS4A protein expression and sTREM2 conflict with Cruchaga’s. In Lisbon, Alison Goate of the Icahn School of Medicine at Mount Sinai, New York, described a single-nucleotide polymorphism in the MS4A locus that removes a repressive element, unleashing MS4A4A and MS4A6A expression. This SNP is a risk allele for AD, hinting that too much of these proteins could be harmful, not protective. What gives? Cruchaga noted that an eQTL for MS4A4A seems to nudge expression in opposite directions in blood and brain samples, which may explain some of the contradictory data. He is currently looking at what this eQTL does in brain-specific cell types to pin down a more definitive answer. Despite the conflicting data, both studies agree that MS4A variants that protect against AD raise sTREM2 levels.
Other research takes a closer look at the effect of sTREM2 on disease. The shedded protein rises in CSF during aging as well as in the prodromal stages of AD, and its levels correlate with CSF p-tau and t-tau (Mar 2016 news). Is sTREM2 beneficial or harmful? Haass believes it’s the former. Michael Ewers, Nicolai Franzmeier, and Marc Suárez-Calvet in his group compared ADNI data from 100 healthy controls with that of 285 people with amyloid and tau accumulation in their brains. Among the latter, 35 people were cognitively normal, 184 were mildly impaired, and 66 had dementia. Regardless of clinical diagnosis, people with AD pathology whose baseline CSF sTREM2 was high declined more slowly on memory and global cognition tests over four years. Also, their hippocampi shrank more slowly, compared with people with low CSF sTREM2 at baseline. Moreover, a high ratio of sTREM2 over p-tau181 correlated with slower progression to cognitive impairment or dementia (Ewers et al., 2019, in press). “I have a feeling that sTREM2 reflects protective microglial activation around the time tangles appear,” Haass said in Lisbon. He told Alzforum that sTREM2 itself may not be the key factor, though. Instead, sTREM2 may be a marker for successful signaling through full-length, membrane-bound TREM2.
While most talks in Lisbon investigated microglia for how they responded to amyloid, Andrea Crotti of Biogen in Cambridge, Massachusetts, is looking at how they handle tau pathology. After ApoE, BIN1 has the largest association with late-onset AD, and it is also expressed in microglia. This protein regulates membrane curvature and endocytosis. Because a previous study found that the p-tau181 in cerebrospinal fluid was hitched to exosomes, Crotti wondered if BIN1 might play a role in the spreading of pathologic tau from cell to cell (Saman et al., 2011). To investigate, he purified exosomes from human CSF, and found that three of 10 patient samples contained exosomes with phosphorylated or oligomeric tau that seeded aggregates in cell culture. Electron microscopy spotted BIN1 on the surface of these vesicles and, lo and behold, revealed that it was an isoform specifically expressed in microglia.
To look for in vivo effects, Crotti turned to mice. The researchers crossed a conditional knockout lacking microglial BIN1 with tau P301S mice, then injected synthetic tau fibrils into the brains of the offspring to accelerate tangle formation. Ninety days later, the BIN1 knockouts had but half as much phosphorylated tau in exosomes, and half as much staining for phosphorylated tau in hippocampal sections as the P301S mice did, leading him to conclude that microglial BIN1 promotes the spreading of pathologic tau via vesicles.
Previous studies have linked BIN1 expression with tangles and tau toxicity (Aug 2012 conference news; Holler et al., 2014; Apr 2015 conference news). These studies did not distinguish between neuronal and microglial BIN1, and at times generated conflicting findings regarding whether BIN1 alters amyloid or tau, once again highlighting the importance of dissecting out the microglial contribution to AD (Apr 2015 conference news).—Madolyn Bowman Rogers
Could CD33 Be the Microglial Target for Stimulating Phagocytosis?
Much of the research on microglia in Alzheimer’s disease has focused on the TREM2 receptor (see Part 4 of this series). But other microglial receptors play a hand in age- and injury-related activation as well (e.g., Apr 2019 news on CD22). One is CD33, a known AD gene that encodes another transmembrane receptor on these cells. There is a protective CD33 variant, and expression level and alternative splicing of the protein affect pathogenicity (e.g., Katsumata et al., 2019). Also known as Siglec-3, CD33 opposes TREM2 signaling and inhibits phagocytosis (Aug 2013 news; Griciuc et al., 2013).
At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, Eloise Hudry of Massachusetts General Hospital, Boston, showcased experiments asking whether suppressing the receptor would unleash phagocytosis and thereby lessen pathology. She knocked down CD33 expression by a third in APP/PS1 mice by injecting an AAV-encoded microRNA targeting CD33 into the mice’s cerebral ventricles. In 8-month-old mice, which have well-established amyloidosis, CD33 reduction caused no change in plaques and only a modest drop in soluble Aβ42 three months later. In contrast, injections into 2-month-old mice slowed plaque growth, reducing total plaque area in the cortex and hippocampus by about one-quarter eight months later (see image below). Along with this, the researchers measured lower levels of some inflammatory markers, such as TNFα, IL1β, and TLR4. The results suggest microglia may have shifted from an inflammatory to a more phagocytic state, Hudry said.
Curb CD33, Curb Plaque? Knocking down the microglial receptor (right) for eight months nudged amyloid plaque downward (red) compared with controls (left). Viral vector, green; nuclei, blue. [Courtesy of Eloise Hudry.]
Another presentation at AD/PD supported the idea that targeting CD33 might facilitate microglial phagocytosis. The protective variant of CD33 lacks a key effector domain that binds sialic acids and triggers intracellular signaling (Malik et al., 2013). Heyne (Cecilia) Lee of AbbVie in Ludwigshafen, Germany, used CRISPR to modify the CD33 gene in microglia made from human iPSCs. In some lines, the scientists cut out only this sialic acid-binding domain (D2-CD33), and in others they knocked out the entire gene. As expected, both cell lines phagocytosed more fluorescent zymosan particles (a yeast glycan) than wild-type microglia did. Intriguingly, while knockouts ate up twice as many particles as wild-types, D2-CD33 microglia did even better, consuming three times as many as wild-type. It is unclear why. A separate experiment lowering the amount of CD33 on the microglial surface also improved phagocytosis, underscoring the idea that targeting CD33 could be therapeutic (Zhao 2018).
However, a recent paper complicates the CD33 story. Researchers led by Steven Estus at the University of Kentucky, Lexington, examined a moderately rare, four-base-pair deletion that truncates the CD33 protein. This results in a complete loss of function in homozygotes and reduced expression in heterozygote carriers. In the IGAP database of 21,982 AD cases and 41,944 controls, the authors found that this deletion had no effect on AD risk. Estus told Alzforum that other unpublished genetic studies support this finding.
To Estus and colleagues, this implies that the D2-CD33 variant may protect not via loss of function, but rather via a gain of function in this receptor. Perhaps the shape of D2-CD33 allows it to bind intracellular activators rather than suppressors, in this way stimulating phagocytosis, Estus suggested. Specifically, the authors believe that instead of its canonical signaling through the SHP-1 adaptor, the mutant CD33 might bind Syk, allowing it to activate the same signaling pathway that TREM2 normally uses, thus enhancing phagocytosis (Estus et al., 2019). Intriguingly, this hypothesis dovetails with the in vitro findings from the AbbVie researchers.
In Lisbon, Peter St George-Hyslop of the University of Toronto, Canada, detailed how the risk and protective variants of CD33 interact. His group crystallized the extracellular domain of CD33 and determined its molecular structure. He found that CD33 forms dimers. The protective variant leads to alternative splicing, creating the form of CD33 that lacks the sialic acid binding site, while the normal variant produces a long protein containing this domain. Dimers of the long form are preferentially trafficking to the cell surface, explaining why these variants have a dominant effect, St George-Hyslop said. He believes the distinct shape of long-form dimers would allow them to bind large polysialylated ligands, and is currently trying to identify those ligands.
Some researchers are exploring the feasibility of targeting CD33. Luisa Quinti, working with Rudolph Tanzi at Massachusetts General Hospital, Boston, developed an assay using an immortalized mouse microglial cell line that expresses human CD33. She selected 37 drugs that previously had been found to affect microglia, and screened them for their ability to alter Aβ uptake and cytokine release in the cell line. Several drugs affected both; Quinti is now examining whether they act through CD33.
She also tested 23 antibodies against human CD33, and found seven that bound CD33 and dose-dependently suppressed its protein level. The assay may be a tool for finding CD33 modulators, Quinti concluded.—Madolyn Bowman Rogers
Could Greasing the Wheels of Lipid Processing Treat Alzheimer’s?
ApoE4 impairs how the brain handles lipids. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, researchers argued that this effect of AD’s biggest risk gene is important in the pathogenesis of late-onset Alzheimer’s. Speakers linked harmful variants of both ApoE and TREM2 to impaired lipid processing, particularly in glia. Conversely, making ApoE “fattier” lowered amyloidosis in disease models, hinting that targeting lipids could have therapeutic potential.
Scientists have long known that ApoE binds lipids and that, in the healthy brain, it is produced primarily by astrocytes. During neurodegeneration, however, microglia pump out ApoE too, Julia TCW of Icahn School of Medicine at Mount Sinai, New York, told the audience. Previously, TCW had reported that lipid processing goes haywire in glial cells carrying the E4 allele. ApoE4/4 microglia and astrocytes generated from human iPS cells dialed up genes for lipid synthesis, while suppressing genes for lipid transport and degradation. This combination caused cholesterol to accumulate in glial cells (Jul 2018 conference news).
In Lisbon, TCW fleshed out her finding with new in vitro data. Comparing E4/4 and E3/3 isogenic astrocyte lines, she found that cholesterol built up only in the former. Much of the astrocytes’ excess cholesterol got stored as cholesterol esters in lipid droplets, though total cholesterol and free cholesterol were also up in E4/4 cells. In healthy glia, ApoE ferried cholesterol from the cell, but the E4/4s made less ApoE protein and allowed less of it out than did E3/3s.
Fat Buildup. Homozygous human ApoE4 astrocytes (right) accumulate more free cholesterol (white) than do homozygous E3s (left). [Courtesy of Julia TCW, Nina Pipalia, and Frederick Maxfield.]
When TCW performed a pulse-chase experiment with low-density lipoproteins, she found that E4/4 astrocytes also took up less lipid from outside than did E3/3s. She traced this to a lack of LDL receptors on the E4/4 glia. “ApoE4 decouples lipid metabolism in microglia and astrocytes,” TCW concluded. In other words, production and clearance become unbalanced, resulting in accumulation of intracellular fats.
ApoE is far from the only gene required for correct lipid processing. In Lisbon, Alicia Nugent of Denali Therapeutics in South San Francisco made a case that the microglial receptor TREM2 plays a similar role. Extracellular lipids are known to bind TREM2 and activate signaling that helps these cells survive, respond to injury, and mop up debris. Notably, this function is disrupted by pathogenic mutations in TREM2 that raise the risk for late-onset AD. R47H and R62H weaken lipid binding, while H157Y speeds shedding of the extracellular portion of TREM2, abolishing intracellular signaling (Feb 2015 news; Kober et al, 2016; Aug 2017 news).
Nugent said that complete absence of TREM2 is even worse, preventing microglia from responding to lipid damage. In wild-type mice poisoned with cuprizone, an agent scientists use to model aspects of multiple sclerosis, oligodendrocytes die and axon tracts in the brain become demyelinated. Microglia clean up myelin debris and, once the toxin is gone, new myelin sheaths form. In TREM2 knockouts, however, remyelination is perturbed and axons degenerate (Poliani et al., 2015).
Why might this be? Nugent and colleagues fed cuprizone to wild-type or TREM2 knockout mice for 12 weeks, then isolated microglia from their brains by fluorescence-activated cell sorting (FACS), and analyzed gene expression. In wild-type microglia, genes for lipid metabolism and lysosomal degradation revved up. This included ApoE, which regulates cholesterol intake and efflux.
The analysis suggested that wild-type microglia challenged by cuprizone ramp up cholesterol metabolism pathways. To do that, the cells enter an activation state reminiscent of disease-associated microglia (DAM) in AD mice, Nugent said (Jun 2017 news). In TREM2 knockouts, this did not happen, indicating these cells cannot initiate the transcriptional programs that are needed for a healthy microglial response to injury.
The researchers confirmed these findings by analyzing the lipid profiles of brain slices from wild-type and TREM2 knockout mice. In the knockouts, cholesteryl esters, the storage form of cholesterol, rose during cuprizone treatment. Other tissue lipids were up as well, including bis(monoacylglycero)phosphate and gangliosides, suggesting a defect in lysosomal degradation of lipids. Lipids accumulated specifically in microglia, not in astrocytes. Nor did the researchers find accumulation in cerebrospinal fluid, which serves as a proxy for the brain’s extracellular space. Intriguingly, TREM2 knockout microglia still took up myelin debris, but they failed to clear it from their cell bodies, resulting in the buildup of cholesteryl esters stored in lipid droplets.
Why did the Denali researchers find lipid accumulation only in microglia, while TCW saw it in both microglia and astrocytes? TCW noted that TREM2 is expressed only in microglia, and thus manipulating it should change only these cells. ApoE, on the other hand, is highly expressed in astrocytes.
Nugent noted that Alois Alzheimer first reported seeing lipid inclusions in glial cells, and others since then have found high levels of cholesteryl esters in AD brain and mouse models (Chan et al., 2012; Morel et al., 2013). TREM2 regulates microglial cholesterol metabolism, and without it, microglia cannot process lipids properly, Nugent concluded. In answer to an audience question, she said it’s unclear if disrupted lipid metabolism is a cause of AD or a consequence.
Could improving lipid metabolism alleviate pathology? The enzyme acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1) converts free cholesterol into cholesteryl esters. When the researchers inhibited ACAT1 in macrophages from TREM2 knockouts, cholesteryl esters did not accumulate. Other studies have found that ACAT1 inhibition lowered both amyloid and tau pathology in AD mice (Puglielli et al., 2001; Hutter-Paier et al., 2004; Bryleva et al., 2010; Shibuya et al., 2015). This line of work did not advance to human trials.
Lipidation, Please. ApoE needs to be properly lipidated to clear Aβ. The E4 allele and aggregated Aβ interfere with this, while “good” cholesterol facilitates it. [Courtesy of Ling Li.]
Some researchers are tackling the problem of blocked lipid metabolism from the other end, looking to speed clearance in the periphery rather than the central nervous system. Peripheral apolipoprotein A-1 (ApoA1) is the main protein component of high-density lipoprotein, the so-called “good” cholesterol. HDL pulls cholesterol from tissues and ferries it to the liver for disposal. Cheryl Wellington of the University of British Columbia in Vancouver, Canada, is using tissue engineering and a reactor system with circulating fluid to build a model of cerebral blood vessels. This allows her to measure trafficking of molecules across the blood-brain barrier. With this system, she previously showed that HDL speeds the flushing of Aβ42 from brain, and can even compensate for ApoE4’s harmful effects on clearance (Oct 2017 news).
In Lisbon, Wellington showed that HDL helps prevent Aβ42 from sticking to collagen and becoming trapped in vessel walls as cerebral amyloid angiopathy (CAA). HDL also suppresses the inflammation of vascular endothelial cells caused by Aβ. HDL levels fall during aging, perhaps contributing to the development of AD, Wellington said.
Peptide Mimetic. 4F mimics the structure of HDL cholesterol, but enters the brain more readily. [Courtesy of Ling Li.]
Could increasing HDL help prevent AD? Ling Li of the University of Minnesota, Minneapolis, studies this question. Li previously found that overexpressing ApoA1 in APP/PS1 mice halved CAA and astrogliosis and restored learning and memory (Oct 2010 news). However, ApoA1 is impractical as a therapeutic, in part because it is large and expensive to make, Li noted.
As an alternative, cardiovascular researchers synthesized an 18-amino acid peptide that mimics HDL. Nicknamed 4F after its four phenylalanines, this mimetic forms amphipathic helices similar to those of ApoA-1. Made from D-amino acids to resist digestion, 4F has undergone a small amount of testing for cardiovascular disease (Navab et al., 2006; Van Lenten et al., 2009; Sherman et al., 2010; Dunbar et al., 2017).
Could 4F be a preventive treatment for AD? Li found that 4F passes through the blood-brain barrier more easily than ApoA1 does, achieving 500 times the concentration of ApoA1 in several mouse-brain regions. In primary astrocyte cultures from mice and human iPSCs, 4F enhanced ApoE secretion and lipidation. The molecule also countered the inhibitory effect of aggregated Aβ42 on ApoE secretion (Chernick et al., 2018; Wang and Zhu, 2018).
In more recent work, Li and colleagues found that 4F treatment changed the lipidation of ApoE4 to that of ApoE2 in primary astrocytes from transgenic mice. To explore 4F in a more complex system, the researchers made brain organoids from human iPSCs (Aug 2013 news; Lindborg et al., 2016). As it did in cell culture, 4F treatment promoted ApoE secretion. In addition, it dose-dependently inhibited Aβ42 aggregation. In a cellular model of the blood-brain barrier, 4F doubled Aβ42 efflux, Li reported in Lisbon.
These data led Li to conclude that HDL-based therapeutic strategies might protect against AD. She noted that the findings once again support the maxim, “What’s good for the heart is good for the brain.”—Madolyn Bowman Rogers
Chimeric Mice: Can They Model Human Microglial Responses?
Microglia are finicky. Take them out of their normal environment and they have a personality meltdown. They dramatically change gene expression and just about turn into a different type of cell. How, then, to study human microglia in a physiological setting? Enter the chimeric mouse. At this year’s AD/PD meeting March 27–31 in Lisbon, Portugal, Bart de Strooper, U.K. Dementia Research Institute at University College London, and Mathew Blurton-Jones, University of California Irvine, described how both their labs independently characterized human microglia grown inside the mouse brain. There, the cells thrive, apparently maintaining their human identity. They “tile” across the brain as normal, yet respond to stress differently than mouse microglia. “With this type of approach, we can begin to ask important questions about the function of human microglia and better understand how they interact with amyloid and tau pathology over time,” Blurton-Jones said.
Scientists have long studied microglia in mice. Alas, while mouse and human microglia have similar transcriptional signatures initially, that changes with age (Jul 2017 news). Most recently, in a paper posted on bioRχiv on April 19, scientists led by Brad Friedman and David Hansen at Genentech, South San Francisco, report that human microglia from AD brains display a signature that differs considerably from disease-associated signatures seen in mice (Srinivasan et al., 2019).
In Lisbon, Renzo Mancuso from De Strooper’s lab at KU Leuven, Belgium, reported that, out of 39 AD risk genes identified in genome-wide association studies, eight, including the microglial receptors CR1 and CD33, have no clear mouse ortholog. Neither do 12 of 43 other genes linked to AD. For another 10 genes, including the microglial receptor and AD risk gene TREM2, the similarity between the mouse and human versions is low. Blurton-Jones agreed. “This is a major problem that makes it difficult to address important questions about the effects of AD risk genes on microglial function using traditional mouse models,” he said.
Chimeric Mouse. Two months after transplanting progenitors into mouse pups, human P2RY12-positive microglia had dispersed throughout the forebrain. [Courtesy of Morgan Coburn and Mathew Blurton Jones.]
Given these differences, researchers have turned to chimeric models. The strategy is to inject the brains of young mice with either human hematopoietic stem cells, or with microglia derived from human embryonic or induced pluripotent stem cells (Abud et al., 2017; Capotondo et al., 2017; Bennett et al., 2018). At AD/PD, De Strooper and Blurton-Jones characterized such transplants, including their transcriptional profiles and responses to Aβ and other forms of stress.
To deplete endogenous microglia, Mancuso treated pups with the colony stimulating factor 1 blocker BLZ945. CSF1 is an essential trophic factor for microglia, and BLZ945 halved their numbers. A day later, he injected human microglia derived from embryonic stem cells into the animals’ brains. The mice were Rag2- and IL2rγ-negative to preclude rejection of the human cells, and they expressed a humanized form of CSF1, since human microglia do not respond to mouse CSF1 (Rathinam et al., 2011). Eight weeks later, the human cells had taken on a seemingly normal, ramified appearance and distributed across the mouse brain in a classic microglial tiling pattern.
Even so, they kept their human transcriptome profile. Mancuso correlated expression profiles of thousands of human microglia isolated from mouse brain with profiles from thousands of cells isolated from temporal cortex tissue removed during neurosurgery. “The profiles overlapped completely,” said De Strooper.
Transcriptomes of human cortical microglia clustered into three main types designated homeostatic, cytokine responsive, and activated. The human cells grown in mice clustered in exactly the same way.
Jonathan Hasselmann and Morgan Coburn in Blurton-Jones’ lab took a slightly different approach. They also used immune-deficient, humanized CSF1 mouse pups, but instead of injecting microglia, they transplanted human iPSC-derived hematopoietic progenitor cells into their brain ventricles and overlying cortices. During the normal human development, these progenitors become microglia and other CNS myeloid cells within the brain, including perivascular, meningeal, and choroid plexus macrophages. Two months later, these researchers also saw the human microglia tile across the mouse forebrain, while a smaller number of human brain macrophages lined mouse blood vessels, meninges, and the choroid plexus. About three-quarters of the microglia were human. Their transcriptional signatures mirrored those of microglia freshly isolated from human brain (Jun 2017 news on Gosselin et al., 2017).
Microglial Dynamics. GFP-expressing human microglia tile across the forebrain (left) and extend highly ramified processes (red) indicative of a homeostatic state. When transplanted into immune-deficient 5xFAD mice (middle, right), the human microglia migrate towards Aβ plaques (blue) and upregulate expression of the DAM marker CD9 (red) [Courtesy of Morgan Coburn and Mathew Blurton-Jones.]
How would the cells respond to Aβ? Mancuso injected 5 μL of 10 μM synthetic oligomers into the mouse brain ventricles eight to 10 weeks after transplanting the human microglia. In reaction to this insult, endogenous mouse microglia shifted their transcriptomes sequentially from homeostatic to cytokine responsive, to activated, with the latter partially overlapping with disease-associated microglial (DAM) signatures described previously (Jun 2017 news). Human microglia in the mice underwent a transcriptional transformation as well, but analysis of more than 10,000 orthologous genes indicated poor correlation between human and mouse microglial responses. Of 207 differentially activated genes, 112 were up in human but not mouse microglia; they included GWAS hits BIN1 and PICALM. “We saw that human microglia responded very differently to Aβ oligomers. This emphasizes the need to look specifically at human cells in the context of AD,” said De Strooper.
Blurton-Jones and colleagues took a different approach by breeding their immune-deficient hosts with 5xFAD mice, then transplanted the human cells into the crosses and tested how the human microglia behaved. In mice that accumulated amyloid plaques, the response was robust and highly localized (see image above). Nine months after injecting human cells, microglia that were right next to a plaque became more amoeboid, downregulated homeostatic genes, and upregulated several DAM genes, as determined by RNA-Seq, including HLA-DRB1, CD9, TREM2, and CD11c. Blurton-Jones said that the human chimeric cell signature only partially overlapped with the mouse DAM signature. Of the 221 differentially expressed genes in plaque-associated human microglia, only about 10 percent match those that are up- or downregulated in the mouse DAM signature. However, Blurton-Jones thinks additional work needs to be done to validate these new genes in human tissue. Some differences could come down to methodology.
Still, De Strooper and Blurton-Jones believe that these chimeric models for now are the way to go to study human microglia in model settings. Of the 30 AD-linked genes with no or poor mouse orthologs, Mancuso found 25 expressed in microglia extracted from patients and 23 expressed in human microglia transplanted into mice. “This emphasizes the importance of using human-specific systems to interrogate genotype-phenotype interactions of the GWAS-identified AD risk genes,” De Strooper noted.—Tom Fagan
Spitting, Sniffing: Is This How We Will Dx Parkinson’s?
The quest for a noninvasive diagnostic test is leading Parkinson’s scientists to unlikely places. One is inside the mouth. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases in Lisbon, Portugal, Giorgio Vivacqua, University of Cambridge, U.K., described his search for pathological α-synuclein in saliva. He claimed that an assay that quantifies synuclein species capable of seeding aggregation was able to pick out PD with high accuracy in a small pilot trial. Another is the spot on the back between a person’s shoulder blades. There, researchers are analyzing a distinctive odor reported to cling to people with PD. According to work from the lab of Perdita Barran, a mass spectrometrist at the University of Manchester, U.K., the musty scent arises from compounds that accumulate in sebum, the waxy, lipid-packed secretion of the skin’s sebaceous glands. A mixture of four volatile chemicals found in sebum largely replicated the “PD smell.” Quantifying their concentration allowed the researchers to distinguish healthy people from those with PD. Alas, for both spit and skin tests, much more research lies between these initial results and a certified, widely available test.
Walter Koroshetz, National Institute of Neurological Disorders and Stroke, Bethesda, Maryland, called the saliva test impressive. “These are still small numbers, but they show good specificity,” he told Alzforum.
Tiago Outeiro, who has university appointments in Göttingen, Germany, and Lisbon, called the sebum work exciting. “It will require a lot of additional work to demonstrate whether this has diagnostic or predictive potential, but we need to keep an open mind about new approaches and follow this up, because it may help us diagnose disease in its early stages,” he told Alzforum.
In a plenary talk at AD/PD, Brit Mollenhauer of Paracelsus-Elena-Klinik, Kassel, Germany, showed examples of how the current, largely clinical diagnosis of Parkinsonian disorders can lead to misdiagnoses that worsen patient care and outcomes. She called on more researchers in the field to join the search for biomarkers at the pre-motor stage of those diseases. Much of that work in the PD field focuses on CSF and blood, REM sleep disorder, and unspecific features such as loss of the ability to distinguish odors. Emerging biomarker studies in saliva and sebum are less well known, and as yet the large cohorts being built to search for biomarkers do not all collect saliva and sebum needed to replicate their early work in larger samples.
Biomarkers for the hallmark pathology of PD—aggregates of misfolded α-synuclein—would be invaluable for definitive diagnosis, to track disease progression, and to evaluate the effectiveness of synuclein-targeted therapies.
Despite initial promise, CSF measures of total α-synuclein have not caught on for diagnosing or tracking progression. People with PD have on average lower CSF α-synuclein, but there’s a large overlap between healthy and disease groups, and new data from the longitudinal Parkinson’s Progression Marker Initiative (PPMI) found little change with time. A new tack may be to zero in on misfolded α-synuclein leaching from the brain’s extracellular space into the CSF. Misshapen or aggregated species have been difficult to reliably measure with ELISA, but that is changing with seeded aggregation assays.
Originally developed to detect infectious prions, these assays, aka protein misfolding cyclic amplification (PMCA) or real‐time quaking‐induced conversion (RT-QuIC), rely on the ability of pathological α-synuclein to induce misfolding of native α-synuclein. In seeded aggregation, a minute amount of CSF sample is incubated with a large excess of recombinant α-synuclein. The mixture is put on a shaker for some hours and, in time, misfolded α-synuclein in the sample initiates condensation of the normal protein into amyloid fibrils, which are quantified by monitoring binding of the fluorescent dye Thioflavin T (ThT). Applied to CSF, RT-QuIC for α-syn seeds can distinguish healthy people from those with Parkinson’s disease with greater than 90 percent sensitivity and specificity (Dec 2016 news; Fairfoul et al., 2016; Groveman et al., 2018).
Perhaps surprisingly, saliva, which is easier to collect than CSF, appears to harbor misfolded synuclein, as well, as Vivacqua and others discovered using Western blotting and ELISA (Vivacqua et al., 2016; Kang et al., 2016; Vivacqua et al., 2019). The likely sources are the autonomic nerves that innervate the salivary glands, which in people with PD are dotted with α-syn aggregates (Beach et al., 2010). Currently, biopsies of the submandibular gland (SMG) and skin provide the only way to document α-synuclein pathology in living people, albeit outside the brain. Some of the large PD biomarker cohorts do include biopsy substudies. Alas, while SMG biopsy carries fewer risks than lumbar puncture, it is uncomfortable, generates usable tissue only 65 percent of the time, and requires the expertise of an ear, nose, and throat specialist (Chahine et al., 2018).
Working in Maria-Grazia Spillantini’s lab, Vivacqua applied RT-QuIC analysis to saliva collected from 36 people who had had PD for an average of two years, and 23 age- and sex-matched healthy volunteers. For quantitation, the researchers tallied the lag time before the rise in fluorescence began, the speed of the increase over 60 hours, and the percent increase at 60 hours. They tested each patient sample three times, in triplicate each time, and achieved high inter-replicate and test-retest reproducibility, Vivacqua said.
Spitting Seeds. Saliva from PD patients, but not healthy controls, initiates α-synuclein aggregation, as detected by increasing ThioflavinT fluorescence. [Courtesy of Giorgio Vivacqua.]
Samples that failed to boost fluorescence above a set threshold after 60 hours were deemed negative. Among the healthy controls, 74 percent (17 of 23) were negative, though 30 percent of the PD patients, or 11 of 36, apparently were as well. Three of these negative patients went on to be diagnosed with tauopathies—two with frontotemporal dementia and one with progressive supranuclear palsy—suggesting they initially had been misdiagnosed with PD.
The as-yet unexplained mismatches in Vivacqua’s study should be followed up, Koroshetz told Alzforum. It would be interesting to know if saliva results correlate with presence of synuclein in submandibular glands, which would give ground truth to the RT-QuIC test, and also to know if the saliva aggregation-positive healthy volunteers become symptomatic, whether with parkinsonism or REM sleep disorder, he said.
For aggregation-positive samples, the lag phase was shorter and fluorescence rose faster in cases compared to controls. These measures distinguished healthy from PD with a sensitivity of 79 and 83 percent, respectively, and a specificity of 100 percent, Vivacqua showed. The fluorescence increase, which could be quantitated in both positive and negative samples, was larger in PD patients, but gave a less-precise separation of the groups, with only 60 percent sensitivity and 88 percent specificity. Vivacqua said he had not yet studied how the RT-QuIC results of α-synuclein in saliva correlate with clinical measures. “We think this is promising for molecular diagnosis of PD,” he concluded.
Michael Schlossmacher, Ottawa Hospital Research Institute, Canada, agreed that saliva is an attractive source of PD biofluid for analysis, but cautioned it may pose unique challenges compared with CSF. Saliva production and concentration fluctuate over the course of the day. Spit harbors a host of microbes, as well as digestive enzymes that chew up carbohydrates and some proteins. “The assays need more subjects and proper consideration of these variables, including medication details. On the plus side, the ease of collection and the RT-QuIC assay lends itself to rapid study of more cohorts,” Schlossmacher said.
Whether spit will signal PD before symptoms begin remains to be seen. Thomas Beach, Banner Sun Health Research Institute, Sun City, Arizona, said he finds Lewy-type α-synuclein deposits in submandibular glands at autopsy in only a third of people who die with asymptomatic α-synuclein brain pathology. Perhaps doing RT-QuIC on SMG or skin biopsy tissue would yield more positives, he said.
Saliva may yet prove to be a good marker of progression. At AD/PD, Beach showed data that serial SMG biopsies revealed a fourfold worsening of α-syn aggregates over four years. It will be interesting to see if that is reflected in saliva, Beach told Alzforum.
Vivacqua said that one outstanding challenge is to characterize the seeds in saliva. Could other aggregated proteins, such as tau, also be present there, and interfere with the assay? “We need to address this, because it is possible that other clinical forms of Parkinsonism could have different types of seed in saliva, as they do in CSF,” he said.
Claudio Soto, University of Texas Medical School at Houston, developed the PMCA assay for prions but was not involved in Vivacqua’s study. He told Alzforum that the Cambridge group has made a good start. To commercialize this assay, Soto founded a company, which he said has developed a faster, automated version that will be used to measure seeding in CSF samples from PPMI. Soto’s current focus is developing the assay for blood, but his group is also investigating saliva, tears, and urine, and even extracts of skin.
The Nose Knows
Other groups are also chasing clues coming off the skin. According to a report published March 20 in ACS Central Science, co-author Joy Milne is a super smeller who can distinguish scents better than most people. Her husband, Les, was diagnosed with PD at age 45 in 1986. A nurse, Milne reported that six years earlier, he had developed a musky odor, which disappeared with treatment and returned as his disease advanced (Morgan, 2016). Scientists began to investigate her story and indeed, tested her nose on T-shirts worn by people with and without PD. She could pick out the disease with perfect accuracy.
The odor Milne picked out was not in sweat. It was in sebum, the light yellow, oily secretion of the sebaceous glands that moisturize skin and hair. Excessive sebum secretion is a non-motor symptom of PD, and likely due to changes in the sympathetic nerves that innervate the glands.
Follow Your Nose. Study scheme to identify volatile compounds responsible for distinctive odor associated with PD. [Courtesy of Trivedi et al., 2019.]
In the new study, first author Drupad Trivedi led the effort to identify the compounds that make up the PD odor (Trivedi et al., 2019). The team collected sebum samples by swabbing medical gauze on the upper backs of patients and matched controls, bagging the gauze, and sending it to a central mass spec lab at the University of Manchester. There, scientists heated the sebum samples to release volatile organic compounds, which were separated and identified by gas chromatography (GC)-mass spec.
The investigators used a discovery cohort of 30 cases and controls, and a separate validation cohort of similar size, recruited from 25 NHS clinics across the U.K. They identified four chemicals in the sebum volatilome whose levels told PD from controls with 90 percent accuracy. Three of the compounds, hippuric acid, eicosane, and octadecanal, were all elevated in patients, although only eicosane levels were statistically significant. In some experiments, the chromatography products went to an odor-detection port, where Milne was stationed to smell the separated compounds. She confirmed the PD odor in the chromatography eluate containing the three compounds. The fourth, perillic aldehyde, was significantly lower in PD sebum. A cocktail of the four chemicals, spiked into normal sebum, mimicked the PD smell closely, according to Milne.
“This is a really well-done study,” said Schlossmacher, who is not a part of this research. “It’s intriguing that both eicosane and perillic aldehyde have also been identified in normal human saliva, as the authors note. Some practitioners, including myself, sense that the ‘PD odor’ in late-onset cases may be associated with changes in our patient’s breath, as well.”
Researchers are pursuing volatilome analysis of breath to detect pulmonary disease and lung cancer (Chang et al., 2018; Lawal et al., 2018), but Schlossmacher said he did not know if this had been done for PD yet.
The published work is part of the larger Nose2Diagnose study led by Barran, which began in 2017. Trivedi told Alzforum the Manchester group is scaling up to validate the results in an additional 350 sebum samples. Ultimately, they will collect 2,000 samples to look at various markers from PD sebum, he said.
Human super smellers are rare, but there is a species whose famously fine nose has stood in the service of humanity for centuries. According to press reports, the researchers are working with a medical dog company to train canines to sniff out PD.—Pat McCaffrey and Gabrielle Strobel
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, Baig F, Ruffmann C, Wade-Martins R, Hu MT, Parkkinen L, Green AJ.
Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies.
Ann Clin Transl Neurol. 2016 Oct;3(10):812-818. Epub 2016 Aug 28
PubMed.
ARIAS: Zooming in On Eye-based Biomarkers for Alzheimer’s
Have biomarkers for preclinical Alzheimer’s disease been staring us in the face all along, and we just did not see them? Recent studies report a plethora of changes in the eye as Aβ pathology emerges in the brain. Dwindling retinal blood flow, thinning retinal nerve fibers, and retinal Aβ plaques themselves are but a few of the features scientists have proffered as readily detectable AD biomarkers. As disparate studies mount, so does the need for consensus. Thus far, there is little independent confirmation, much less agreement on what works. Peter Snyder of the University of Rhode Island, Providence, is trying to change that. At the AD/PD meeting in Lisbon, Snyder announced an initiative to compare eye-based measures head-to-head, and longitudinally, in people across the disease continuum. If his effort confirms eye-based tests as bona fide AD biomarkers, they could become frontline screens that could be deployed around the world.
“For those of us doing research in this field, the dream is that universal and regular vision care with retinal imaging could serve to both preserve physical/social functioning in older adults and detect early signs of cognitive disease,” wrote Alison Abraham of Johns Hopkins School of Medicine in Baltimore.
“Eye-based screening could have a meaningful role in future studies of preclinical Alzheimer’s disease even in an era when we have high hopes for fluid and imaging biomarkers,” noted Pierre Tariot of the Banner Alzheimer’s Institute in Phoenix.
Studies have linked numerous eye changes to AD or cognitive impairment. Many focus on the retina, a disc of nervous tissue at the back of the eye that connects to the brain via the optic nerve. PubMed lists around 100 studies published in the past three years about the retina and AD. For example, the retina can be visualized with ocular coherence tomography. Opthalmologists and optometrists use OCT to identify glaucoma or macular degeneration, and angiography recently has been tacked on to add blood flow measurements. This is called OCT-A.
Zoom in on AD? A peripapillary OCT scan images a region (green circle) of the retina surrounding the entrance to the optic nerve. The analysis renders a linearized image in which each retinal layer can be measured. [Courtesy of Peter Snyder.]
Researchers use OCT to detect Aβ plaques in the retina and attempt to correlate them with cortical Aβ accumulation as per PET scans (Jul 2014 conference news; Sep 2017 news). Others link retinal capillaries to AD. For example, one study reported that the inner fovea—an avascular zone in the middle of the retina where visual acuity is highest—was thinner and wider in cognitively normal people with a positive amyloid PET scan (Aug 2018 news). Another claimed slower blood flow and fewer vessels in the surrounding parafoveal region in people with mild cognitive impairment (Zhang et al., 2019). Using photography, a study found double the odds of mild cognitive impairment or dementia in people whose retina had hemorrhaged than in people without such bleeds (Lee et al., 2019).
Synaptic Sandwich. The retina comprises 10 layers, including eight depicted here. Three neuronal layers (GCL, INL, and OPL) are connected via synaptic layers (IPL, OPL). The retinal nerve fiber layer (RNFL) projects axons into the optic nerve. [Courtesy of Peter Snyder.]
The structure of the retina itself is also implicated in AD. It consists of 10 layers, including three neuronal cell layers sandwiched on either side by synaptic and axonal layers. Studies have come to conflicting conclusions about how the thickness of each layer correlates with AD. Some report that people with AD have fewer ganglion cells—the neurons project their axons into the optic nerve like hairs pulled into a ponytail. Others report a thinner retinal nerve fiber layer (RFNL), which contains those optic nerve-bound axons; alas, it’s unclear exactly where along the retina the RNFL thins, if at all (Danesh-Meyer et al., 2006; Cheung et al., 2015; Garcia-Martin et al., 2016). The list of single-study biomarker suggestions goes on (for review, see Chan et al., 2018).
At AD/PD, Snyder reviewed his own data of one of the few longitudinal studies thus far to examine the relationship between multiple retinal measures and preclinical AD (Santos et al., 2018). He also presented unpublished findings from the same cohort. The researchers enrolled 63 people with a family history of AD who were cognitively normal but reported subjective memory complaints. They measured numerous retinal markers in them at baseline and 27 months later, at which time participants had an Aβ-PET scan. Santos et al. spotted a correlation between a person’s Aβ accumulation and thinning of the RNFL in their macula, a pigmented region near the retina’s center. In Lisbon, Snyder added an analysis of macular pigment optical density (MPOD), a measure of pigment within the macula. Using a combination of lasers to detect pigment, Snyder found that reduced pigment correlated with slippage on cognitive tests, but not with Aβ deposition.
Enter ARIAS
Snyder likened the current landscape of eye-based biomarkers to the pre-ADNI days of neuroimaging. Given the jumble of small studies reporting on a group’s “favorite” single marker, Snyder decided to kick off a larger, longer cohort to track multiple candidate markers across the disease spectrum. Called Atlas of Retinal Imaging in Alzheimer’s Study, ARIAS aims to amass a reference database of retinal changes that track with disease (see clinicaltrials.gov). To be funded by Florida’s BayCare Health System, ARIAS will recruit participants from two hospitals in Tampa, Florida, and from one in Rhode Island.
ARIAS seeks 330 participants aged 55 to 80, from cognitively normal people deemed at low or high risk for AD based on family history and ApoE4 status, to people with mild cognitive impairment or mild AD. They will undergo annual eye exams for three years, at which investigators will use OCT and OCT-A to measure the thickness of every retinal layer, blood vessel volume and flow, and macular pigment. They will assess the pupil’s response to light, and sensitivity to contrast, a visual marker that wanes in AD. A subset of participants will wear actigraphy monitors for two weeks to gauge their physical activity and sleep patterns, as connections between the eye and the brain are known to affect sleep. Amyloid PET scans are not in the protocol, but Snyder said many participants have already had one as part of the Imaging Dementia–Evidence for Amyloid Screening (IDEAS) study (see clinicaltrials.gov), and that data can go into ARIAS analyses.
“This longitudinal study in well-characterized (preclinical) AD cases provides a great step forward for retinal imaging as possible biomarker in AD,” commented Jurre den Haan and Philip Scheltens of VU University Medical Center (see full comment below). Longitudinal retinal imaging data from preclinical AD cases from the European Medical Information Framework (EMIF)-AD cohort will soon be published.
Carol Cheung of the Chinese University of Hong Kong believes ARIAS will fill a knowledge gap in the field. Interpreting retinal parameters is tricky because they can be affected by aging, diseases such as hypertension and diabetes, eye size, glaucoma, and other neurological conditions including multiple sclerosis and stroke. “It is likely that AD-related changes in retinal vasculature and neuronal structure present as a spectrum of variants due to involvement of multiple variables,” she wrote.
Maya Koronyo-Hamaoui of Cedars-Sinai Medical Center in Los Angeles agreed that studies such as ARIAS were highly needed to find eye-based biomarkers that could facilitate early AD detection and monitoring of progression. “Moreover, there is a great need to correlate these nonspecific changes in retinal vascular biomarkers and atrophy with retinal AD-characteristic signs (amyloid beta and tauopathy) in these patients,” she added.
Logistical Hurdles
Snyder hopes results from ARIAS will form the foundation for a future toolkit of eye-based biomarkers that can be used as a frontline screening tool for preclinical AD. A person who tests positive for eye-based markers could be referred to a neurologist for more standard biomarker tests. OCT tests could theoretically be run in primary care clinics, but Snyder said that adding one more test to the already packed to-do list of these physicians might not be easy. Opthalmologists also have a loaded schedule and perform a variety of procedures. Optometrists, he said, are more likely to welcome Alzheimer screening into their standard care. Not only do they have fewer tests to run on their patients, they may also see AD screening as a way to ensure a steady client base, Snyder said. OCT scanners are cheaper and more widely available around the world than PET scanners.
Biomarker by Hand? A handheld electroretinogram (ERG) measures the response of retinal neurons to light, without touching or dilating the eye (left). An ERG signal from the device screen shows a loss of the photopic negative response in a person with MCI (right). [Courtesy of Edmund Arthur and Delia Cabrera DeBuc, University of Miami.] [Courtesy of Edmund Arthur and Delia Cabrera DeBuc, University of Miami.]
That said, Delia Cabrera DeBuc of the University of Miami pointed toward even cheaper tools. Last December, she reported that retinal neurons are hypoactive in people with cognitive impairment. She uses a handheld electroretinogram (ERG, see Cabrera DeBuc et al., 2018). The device costs less than $15,000, requires no dilation or manipulation of the eye, and is easier to use than an OCT scanner, which costs around $100,000, Cabrera DeBuc said.
Even with an electroretinogram, Cabrera has seen pushback in primary care settings, where nurses and doctors are crammed for time. She agrees that optometrists’ offices might be a venue for AD screening using OCT, though patients without insurance for vision care would have to pay out of pocket to receive tests there. Therefore, incorporating eye-based AD screening tests into the primary care reimbursement structure would be key to their widespread use, Cabrera DeBuc said. She thinks ARIAS is on the right track, because any retinal AD markers that emerge from it will motivate the health care field to implement screening into primary care.
Snyder is hosting an open workshop May 22–23 in Washington, D.C., where researchers can discuss new data and challenges in the field of retinal imaging, and hash out a way forward. Register by clicking here.
Beyond AD
Eye-based biomarkers could hold promise for detecting other neurodegenerative diseases as well. One study found thinning in the retina’s outer photoreceptor layer in people with frontotemporal dementia (FTD) (Kim et al., 2017). This month, Benjamin Kim at the University of Pennsylvania in Philadelphia and colleagues published longitudinal findings from the same cohort. The outer retina continued to diminish, but only in FTD patients predicted to have underlying tau pathology (Kim et al., 2019). This is distinct from the inner layer thinning observed in AD. “This suggests that OCT may help to distinguish FTD from AD. We currently are enrolling patients in an OCT study to directly compare these two groups,” Kim wrote to Alzforum.
Retinal thinning in the rTg4510 mouse model of tauopathy suggests that changes in the eye reflect tau pathology in the brain (Harrison et al., 2019). Retinal changes have also been reported in people with amyotrophic lateral sclerosis (Volpe et al., 2015).—Jessica Shugart
Despite years of study, ApoE still has secrets to give up. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, researchers indicted ApoE4 on several fronts. They charged it with holding back the microglial response to amyloidosis. They said it bungled disposal of damaged mitochondria, perhaps perturbing cellular energy metabolism. They implicated it in axonal remodeling, and said this may explain why the highly arborized neurons of the entorhinal cortex are particularly vulnerable to Alzheimer’s. Finally, as peripheral ApoE is beginning to attract more attention, scientists are accounting for the different pools of this lipoprotein that course through the human body. Others elaborated on how ApoE4 damages blood vessels in the brain and correlates with neuroinflammation. Quite the rap sheet.
First, microglia, the “phenom” in AD research these days. Many genetic risk factors act through their effect on these cells (see Part 4 of this series), and some appear to be in cahoots with each other. Oleg Butovsky of Brigham and Women’s Hospital, Boston, previously reported that TREM2 and ApoE cooperate to rouse microglia into a “microglial neurodegenerative” (MGnD) state, in which they clean up dead neurons and other debris (Feb 2015 conference news; Sep 2017 news).
Microglial Replacement. ApoE4 microglia freeze into a homeostatic state; could replacing them with ApoE3 monocytes from blood restore normal function? [Courtesy of Oleg Butovsky.]
How does the ApoE4 allele affect this activation? In Lisbon, Butovsky said it has the same effect as knocking out ApoE, i.e., it traps microglia in a homeostatic state. His team isolated microglia from adult mice that carry humanized ApoE3 or ApoE4. In the former, microglia activated as the animal aged, but in the latter, homeostatic gene expression stayed high. Then the scientists challenged microglia by injecting dying neurons into mouse brain. Normally, this spurs microglia to enter the MGnD state. In E4 mice, however, it did not. They managed to turn down their homeostatic genes, but not to ramp up MGnD genes, like a teenager who can’t work up the motivation to get off the sofa and clean his room. Butovsky concluded that ApoE4 microglia are unable to respond properly to cellular damage in the brain.
In addition, aberrant behavior of ApoE4 microglia during development may render the brain vulnerable to Alzheimer’s, Butovsky believes. Charlotte Madore in his group found that microglia in ApoE3 mice assumed an MGnD phenotype during the first two weeks of life, when the cells are busy pruning inactive synapses. Then microglia settle into a calmer mode, turning up homeostatic genes. In newborn ApoE4 mice, however, microglia maintained high expression of homeostatic genes throughout infancy and into adulthood. Madore thinks this may prevent them from appropriately sculpting brain circuitry, and could lead to behavioral abnormalities later in life. Similarly, genetic ablation of TREM2 makes microglia “superhomeostatic” and prevents them from migrating toward tissue damage (May 2017 news). TREM2 knockout mice have abnormal social behavior as adults (Filipello et al., 2018; Calcagno et al., 2018).
Madore is investigating if these microglial differences interact with early life stress. Butovsky, working with Arie Kaffman at Yale University in New Haven, Connecticut, previously reported that early stress can cause lasting changes in microglia gene expression (Delpech et al., 2016). Madore found that stressors such as lack of bedding material cause mouse microglia to ramp up MGnD gene expression. If E4 mice cannot mount this response, perhaps that affects the brain in ways that leave mice susceptible to degeneration later, she speculated. In people, childhood epilepsy in ApoE4 carriers has been linked to an increased risk of AD late in life, hinting that early stressors could interact with genetic factors to predispose someone to the disease, Madore believes (Joutsa et al., 2017).
If ApoE4 microglia make the brain less able to combat AD, perhaps replacing them with healthier cells could be therapeutic, Butovsky suggested. In other studies, depleting brain microglia stimulated blood monocytes to enter the brain and repopulate the microglial niche (Lund et al., 2018; Cronk et al., 2018; Bennett et al., 2018). Microglia numbers can be reduced by preventing their proliferation with CSF1R blockers. Such drugs are under development for cancer, and treatment with one lessened pathology in a mouse model of AD (Sosna et al., 2018; Cannarile et al., 2017). Butovsky suggested infusing healthy ApoE3 monocytes into the bloodstream before CSF1R inhibitor treatment, to allow these cells to move into the brain and take over immune surveillance duties there. He is testing this idea in mouse models, collaborating with Li-Huei Tsai at MIT and Mathew Blurton-Jones at the University of California, Irvine.
ApoE affects other cellular processes that could play a role in AD, as well. Shira Simonovitch of Tel Aviv University, Israel, working with Daniel Michaelson and Ronit Pinkas-Kramarski there, previously reported that ApoE4 suppresses autophagy (Simonovitch et al., 2016). In Lisbon, Simonovitch extended this finding to mitochondrial disposal. The cell’s energy powerhouses are dynamic organelles that fuse and divide in response to stressors. Fusion boosts their health, while fission allows malfunctioning mitochondria to be chewed up by autophagosomes, a process known as mitophagy.
Simonovitch compared this in transgenic mice carrying human ApoE3 or ApoE4. In hippocampal sections from the E4 mice, Simonovitch counted three times as many elongated mitochondria as in E3 hippocampus. This hints at a block in fission, and staining for specific proteins revealed a dearth of the fission protein Drp-1 and an excess of the fusion protein Mfn1.
The scientists also saw that mitochondria in E4 brains had fewer crista, the folds of the inner membrane that produce energy, and western blots indicated changes in several known mitochondrial proteins. In particular, E4 mitochondria had a shortage of the cleaved form of pink1, which is associated with healthy mitochondrial resting potential. The data hint at an energy deficit in the ApoE4 brain; the increased mitochondrial fusion may be an attempt to compensate, Simonovitch suggested.
How about humans? The researchers have no way of measuring this directly in living people, but they compared postmortem samples from the brains of ApoE4/4 and ApoE3/3 AD patients. They found half as much Drp-1 in the former as the latter by western blot. Impairments in mitophagy have been linked to Parkinson’s and other neurodegenerative conditions (Aug 2013 news; Oct 2014 news; Sep 2016 news). Mitochondrial damage occurs in AD models, and removing these damaged organelles lessens pathology and improves memory in mouse and worm models of AD (Nov 2009 news; Feb 2010 news; Feb 2019 news).
Axonal Vulnerability? Entorhinal cortex neurons possess enormous axonal arbors (bottom right) that may leave them susceptible to tau pathology. [Courtesy of Tamamaki and Nojyo, 1993.]
ApoE4 also promotes tau pathology (Sep 2017 news). Neurons in the layer II entorhinal cortex (EC) and hippocampal CA1 region are particularly vulnerable to tau tangles, and Jean-Pierre Roussarie and colleagues at Rockefeller University wondered if ApoE isoforms might influence this. Roussarie works in the laboratory of Nobel laureate Paul Greengard, who passed away April 13 at age 93 (see obituary). Roussarie profiled seven different neuronal types in wild-type mice at three different ages to identify pathways most altered in vulnerable neurons compared with those that resist tau pathology. Together with Olga Troyanskaya’s group at Princeton University, Roussarie and colleagues then combined these expression profiles with a compendium of human genomics and GWAS data for genes associated with tau pathology (Greene et al., 2015; Beecham et al., 2014).
This pinpointed a tau vulnerability gene module that was highly connected in EC neurons, and whose gene members were suppressed during aging and in APPswe/PS1dE9 mice. Genes in this cluster regulate axon extension, microtubule assembly and transport, and neuronal signaling (Brichta et al., 2015; Roussarie et al., 2018). Importantly, gene-expression profiles from the different neuron types were comparable to those in postmortem human brain samples from the same regions, suggesting the results may translate to people, Roussarie said. This work is described in a paper posted to bioRχiv (Roussarie et al., 2018).
How about ApoE? Roussarie compared gene expression in healthy, year-old female ApoE2 and ApoE4 mice, and found 57 neuronal genes that were differently expressed in the E4s, about half turned up and the other half down. These genes overlapped extensively with the tau vulnerability module of axonal plasticity genes, as well as with genes associated with tau tangles in another GWAS (Chibnik et al., 2017). In other words, ApoE4 affects the same set of genes as do aging and AD pathology. Within EC neurons, the effects of ApoE4, aging, and tau pathology all converge on axonal plasticity, Roussarie concluded. He noted that EC neurons extend extremely complex axonal arbors, and speculated that this may leave them vulnerable to perturbations in this process.
While most studies of ApoE focus on the central nervous system, the protein is also abundant in the periphery. In Lisbon, Randy Bateman of Washington University in St. Louis reminded the audience that CNS and PNS versions are processed differently. In the CNS, all three isoforms turn over at the same rate, while in blood, they vary greatly. There, ApoE4 is cleared twice as fast as ApoE3. In line with this, people with one E3 and one E4 allele have more E3 than E4 in their plasma, while the opposite is true in brain (Wildsmith et al., 2012; Baker-Nigh et al., 2016).
To examine brain and plasma ApoE more closely, since then Bateman and colleagues have developed an assay to isolate ApoE from body fluids via monoclonal antibodies and analyze the protein by mass spectrometry. This taught them that, in cerebrospinal fluid, ApoE’s C-terminal domain was either missing or modified, Bateman reported in Lisbon. In addition, plasma and CSF ApoE differed at threonine 194. Though the researchers are still determining what this modification is, there are previously reported candidates at this location (Wernette-Hammond et al., 1989; Halim et al., 2013). These differences may help researchers design therapies to specifically target pathological ApoE, Bateman suggested.
Other research implicates peripheral ApoE in AD. Guojun Bu of the Mayo Clinic in Jacksonville, Florida, previously reported that peripheral ApoE4 makes brain blood vessels leaky and slows cerebral blood flow. This correlated with worse memory and more amyloid plaques in brain, even in animals without any CNS ApoE (Jul 2018 conference news). In Lisbon, Bu added to these data, reporting that blood vessels in mice with peripheral ApoE4 branch less than those in E3s and dilate less frequently, suggesting problems with regulating blood flow. E4s also deposited less collagen around blood vessels and had more astrogliosis in their brains.
To confirm that the peripheral ApoE4 effects also occur in wild-type animals that have CNS ApoE, Bu’s team injected plasma from young ApoE4 mice into aged wild-type mice, and found that it promoted leakage of the blood-brain barrier. ApoE3 plasma had the opposite effect, stemming leaks. “ApoE4’s harm likely comes from a combination of central and peripheral effects,” Bu concluded.—Madolyn Bowman Rogers
Off-Balance Endocytosis Lays Groundwork for Disease
Endocytosis is implicated in Alzheimer’s pathogenesis, because the continuous swallowing up of proteins from the cell surface and packaging them into little organelles brings the amyloid precursor protein (APP) into contact with the secretases that chop it and produce Aβ. How this changes as cells age, though, and how genetic risk factors for Alzheimer’s influence it, is still unclear. Data presented at the14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, suggest that as cells get older, the endocytosis of APP speeds up, giving it more access to its cleaving enzymes and increasing its processing. Other presentations showed that over- or underexpressing certain endosomal factors brings on symptoms of disease, suggesting that too much or too little endocytosis could kick off intracellular disease before pathology accumulates.
Faster with Age
Many scientists believe that sluggish Aβ clearance in aging is a cause for late-onset AD (LOAD) (e.g., Saido and Leissring, 2012; Aug 2016 conference news). This stands in contrast to the increased Aβ production responsible for autosomal-dominant forms of the disease caused by mutations in APP or γ-secretase. Could a similar revving up of APP processing and Aβ production also come into play in LOAD, and if so, does endocytosis have something to do with it?
To find out, first author Tatiana Burrinha and colleagues in the lab of Claudia Almeida, University Institute of Lisbon, Portugal, cultured primary embryonic cortical neurons from wild-type mice and aged them. These neurons undergo three differentiation stages in a dish: They develop axons and dendrites after a week, reach peak maturation at 21 days, and exhibit signs of aging at 28 days, including synapse loss and increased free radicals. The researchers compared 21-day-old mature neurons with 28-day-old ones to look for differences in Aβ levels or APP processing.
More APP with Age. As cultured neurons get older, their neurites endocytose more APP, leading to more Aβ production. [Image courtesy of Tatiana Burrinha.]
In the old neurons, immunofluorescence using anti-Aβ42 antibody 12F4 revealed 50 percent more intracellular Aβ42 specifically in neurites. There, early endosomes were larger than in younger neurites. Western blots with the anti-APP APPY188 antibody indicated that intracellular APP C-terminal fragments (CTFs) doubled in aged cells compared with mature cells, but full-length APP remained the same, suggesting its expression did not change. Old cells had half the number of pre- and postsynaptic markers as 21-day-old cells, and a γ-secretase inhibitor rescued about 40 percent of that deficit. Together the results hinted that the trafficking of APP changes with aging, and this harms synapses.
To see if these observations held in vivo, the researchers compared the levels of processed APP fragments between 18-month aged wild-type mice and 6-month-old young adults. According to western blots on cortical homogenates, α- and β-CTF fragments doubled in the aged animals relative to young adults. Levels of the EEA1 marker of early endosomes tripled, while the amount of η–CTF rose as well. Together, the data suggest that APP processing goes up with aging in vivo, said Almeida.
That synapses decline in these experiments suggested to the authors that the age-related increase in Aβ production is a cause of synapse loss in age-related disease. It may add to synaptoxicity caused by previously reported defects in the clearance of Aβ42, said Almeida (Iwata et al., 2002; Solé-Domènech et al., 2016; Elahy et al., 2015).
Almeida’s research doesn’t point to a specific drug target yet, but could in the future. “Once we know how APP endocytosis is increased with neuronal aging we may develop an inhibitor that does not interfere with synaptic vesicle endocytosis,” said Almeida. “This drug could potentially prevent amyloid accumulation and synaptic decline with aging.”
Gopal Thinakaran, University of Chicago, cautioned that data from aged cells in a dish might not represent what is going on in the aging brain. Lotta Agholme, University of Gothenburg, Sweden, echoed his concern. “Just neurons in a dish is a tricky thing, because you don’t have the whole brain. If you don’t have Alzheimer’s-related processes going on, it’s hard to find a good model,” Ralph Nixon, New York University, said. “Aging in vivo is more involved than neurons that have been in a dish for weeks.”
That said, the in vivo data is a step toward validating the result, the commentators agreed. Nixon lauded that Almeida analyzed APP processing fragments besides Aβ. He believes β-CTFs are toxic in their own right. At AD/PD, Eunju Im from Nixon’s lab showed that the elevated levels of β-CTFs in fibroblasts from people with Down’s syndrome and human APP-expressing N2a cells interfered with lysosomal degradation, impeding assembly of the complex on the lysosome surface necessary to render these organelles sufficiently acidic. Elevating β-CTFs in 2N human fibroblasts by inhibiting γ-secretase led to similar deficits.
Mice Model Endosomal Problems
Nixon presented postdoctoral fellow Anna Pensalfini’s data on a new mouse model of neuronal Rab5 overactivation that leads to increased endocytosis and enlarged endosomes in AD. Rab5 is a GTPase that helps initiate endocytosis and fuse the early endosomes that make sorting and signaling endosomes. APP’s β-CTF binds and activates Rab5, hyperactivating it in AD. However, in this model, Rab5 was expressed and activated to comparable levels as in AD and Down’s syndrome, without added APP fragments.
At 7 months, Rab5T mice developed upregulated endocytosis, enlarged endosomes, enhanced long-term depression, reduced long-term potentiation, shorter, sparser dendritic spines in the hippocampus, and tau hyperphosphorylation. The mice floundered on the novel object recognition test and their basal forebrain cholinergic neurons degenerated at 11 months.
“This argues strongly that endocytosis specifically as activated by Rab5 is capable of triggering a cascade of biological events that phenocopy synaptic and cholinergic deficits seen in AD,” Nixon told Alzforum.
For his part, Thinakaran introduced a conditional BIN1 knockout mouse. BIN1 is a common risk factor for Alzheimer’s disease, according to genome-wide association studies. Scientists don’t know how BIN1 normally functions in the brain. Some suspect it regulates BACE trafficking, and too little of it appears to hasten tau propagation (Nov 2015 news; Oct 2016 news). Earlier this year, Thinakaran’s lab reported that reduced BIN1 expression affected neither Aβ levels nor BACE1 localization (Andrew et al., 2019).
In Lisbon, Thinakaran reported that synaptic transmission was weaker in hippocampal slices of mice lacking BIN1 in excitatory neurons than controls. And while high-frequency stimulation depleted vesicles in controls within five to 10 seconds, BIN1-less neurons sustained synaptic vesicle release for far longer. Using super-resolution microscopy, postdoctoral fellow Pierre De Rossi saw that without BIN1, SNARE proteins that normally facilitate fusion of plasma membranes and vesicle release were disorganized. Using three-dimensional reconstruction of CA3 to CA1 synapses, the researchers saw more synaptic vesicles either docked or hanging around in the reserve pool, which suggests fewer were successfully released. In addition, the cKO neurons were endocytosing more and making more vesicles. The mice struggled to find the hidden platform in the Morris water maze.
“Different lines of data are consistent with each other,” Thinakaran told Alzforum. “They all point to a role for BIN1 in presynaptic vesicle release that has a profound impact on memory consolidation.” Changes in BIN1 localization or levels could affect synaptic transmission and cognition independent of any pathology, he added.—Gwyneth Dickey Zakaib
Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
As neuroinflammation becomes ever more deeply implicated in Alzheimer’s disease, therapies targeting this process are starting to enter clinical trials. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, speakers presented several approaches under investigation. Two groups have generated anti-TREM2 antibodies that they claim activate the receptor and stimulate microglia to remove amyloid. One of these has entered a Phase 1 trial. One company is taking an antibody against CD33 into human studies, while another group is investigating an ApoE antibody in mice. Meanwhile, a small molecule that started out being tried in cardiovascular disease a decade ago turns out to douse inflammation via an epigenetic mechanism. Could it boost cognition in aging?
It’s too early to tell if any of these approaches will work. Even so, at a time when Alzheimer’s researchers are finally ready to turn the page on NSAIDs (Apr 2019 news), the buzz in Lisbon reflected their eagerness to start minting their intense interest in inflammation into new therapeutic approaches.
Is TREM2 a Viable Therapeutic Target?
First, the poster child for microglial AD risk. The receptor TREM2 prods microglia to police amyloid plaques, and people with high levels of its soluble portion in their CSF decline more slowly at all stages of Alzheimer’s disease (May 2017 news; Part 4 of this series). These findings would appear to make TREM2 an attractive target—except therapies would have to boost it. Activators are generally harder to design than inhibitors.
TREM2 Activation. Binding of an antibody (red) to TREM2 can trigger signaling through its co-receptor DAP12, resulting in phosphorylation of Syk. [Courtesy of Donna Wilcock.]
Now, two groups claim to have done it. One is the biotech company Alector, based in South San Francisco, which generated a monoclonal antibody against TREM2. In Lisbon, Donna Wilcock of the University of Kentucky, Lexington, said the mouse version of the antibody, AL002a, binds and activates TREM2. In cell culture, AL002a treatment increased phosphorylation of Syk, a downstream effector of TREM2 signaling.
Wilcock’s group previously reported efficacy for this antibody in APPPS1 mice (Dec 2016 conference news). In Lisbon, she added new in vivo data. The researchers injected 10 μg of AL002a or IgG isotype control into the frontal cortices and hippocampi of 5-month-old 5XFAD mice. Three days later, immunostaining indicated Aβ levels had been halved, while pro-inflammatory and anti-inflammatory cytokines had spiked, suggesting microglial activation.
The researchers then injected 50 μg/kg AL002a into 4-month-old 5XFAD mice intraperitoneally for 14 weeks. After this, treated transgenics had 50 percent more activated CD11b+ microglia than untreated ones, along with elevated pro-inflammatory and anti-inflammatory cytokines. Treatment doubled the number of microglial cells surrounding amyloid plaques, and halved the amount of Aβ. Treated brains harbored fewer diffuse plaques than untreated, but compact amyloid remained in place, Wilcock said.
What about behavior? Mice receiving AL002a recognized new objects and navigated a radial arm water maze as well as wild-types did; untreated transgenics performed poorly. Future studies will examine if AL002a affects tau pathology, and test it in older mice with more advanced pathology, Wilcock said. In answer to audience questions, she said she has seen no adverse vascular effects so far.
The human version of the antibody, AL002, last November entered its first Phase 1 trial for AD patients, conducted at multiple sites in Australia as well as one in London and one in Orlando, Florida. Researchers will infuse a single dose of AL002 or placebo to 51 healthy adults, stepping up the dose after the prior, lower dose appears safe. If volunteers tolerate these single doses well, 16 people with Alzheimer’s will be randomized to multiple doses of AL002 or placebo. Alector is developing AL002 in collaboration with Abbvie.
The Alector antibody is not the only one to activate TREM2. In Lisbon, Christian Haass of the German Center for Neurodegenerative Diseases in Munich described a similar monoclonal antibody his lab has developed against the mouse receptor. He said his antibody also boosts phospho-Syk signaling in cultured cells in a dose-dependent fashion, and that functional assays support the idea that it activates TREM2 signaling. Treatment helped cultured macrophages survive growth-factor deprivation, Haass said. In primary microglial cultures, the antibody stimulated phagocytosis of myelin debris. Haass presented more data at the Federation of European Neuroscience Societies (FENS) conference on AD, held May 5–8 in Rungstedgaard, Denmark.
Others are developing anti-TREM2 antibodies as well. In a conference poster, Hyun Jung Kim of the Korea Brain Research Institute in Daegu described the generation of two different monoclonal antibodies raised in mouse against purified human TREM2. Both antibodies reportedly immunoprecipitated TREM2 from transfected HEK293 cells, forming a stable complex with the receptor. Each antibody recognized a different region of TREM2. The poster did not describe the functional effects of these proteins.
Anti-TREM2 antibodies could perhaps serve as biomarkers, as well as therapeutics. Researchers led by Silvio Meier of Uppsala University, Sweden, are developing a version that could be used for PET imaging. They conjugated an experimental anti-TREM2 monoclonal antibody, mAb1729, to a moiety that binds the transferrin receptor to enable its uptake into brain. They radiolabeled this complex with iodine 125, then injected these bispecific antibodies into 16-month-old Tg-ArcSwe, Tg-Swe, and wild-type mice. One day later, uptake was about 50 percent higher in transgenic mice than in wild-types. This reflected the higher levels of TREM2 in transgenic brain, the researchers found. The tracer eventually could be used to measure microglial activation in AD brain, they suggested.
Other Ways to Tweak Microglial Activation
Of course microglial activation entails far more than TREM2, and researchers are exploring other targets. For example, Alector just began its first trial of the anti-CD33 antibody AL003. The microglial receptor CD33 opposes the effects of TREM2 signaling, and may make a more amenable target because it would be inhibited rather than activated (see Part 5 of this series). In the first phase of the trial, 42 healthy adults will receive a single treatment of either placebo or one of seven different AL003 doses. The second, multiple-dose phase will enroll 12 patients clinically diagnosed with Alzheimer’s, two of whom will receive placebo. The outcomes are to learn about safety and tolerability, and to determine the highest achievable concentration of AL003 in serum and cerebrospinal fluid. The trial is enrolling in Melbourne, Australia.
ApoE, too, is becoming an intriguing target. Amyloid plaques are loaded with this protein, and much of it seems to come from microglia (Jan 2019 news). David Holtzman, Washington University in St. Louis, previously described an antibody, HAE-4, that binds preferentially to aggregated, nonlipidated ApoE. ApoE4 is poorly lipidated, a feature that seems to promote amyloid pathology (Dec 2011 news; May 2014 news; Part 6 of this series). When the researchers injected HAE-4 into APPPS1 mice expressing human ApoE4, it triggered microglial activation and halved plaque load after six weeks (Apr 2018 news).
In Lisbon, Monica Xiong in Holtzman’s group presented new data on this antibody. She noted that many anti-amyloid approaches cause microhemorrhages and swelling in the brain, called amyloid-related imaging abnormalities, or ARIA, particularly in people who have extensive cerebral amyloid angiopathy (CAA). To find out if HAE-4 has this effect, Xiong and colleagues tested it in 5XFAD mice that have human ApoE4. This model develops mostly CAA with a few parenchymal plaques (Liao et al., 2015). The researchers compared HAE-4 to a chimeric mouse version of aducanumab as well as to control antibody. The chimeric antibody conjugates aducanumab to a mouse Fc domain to enable activation of mouse microglia. They administered 50 mg/kg of each antibody weekly for eight weeks, starting two months after plaques formed. The anti-ApoE antibody reduced plaque load by 40 percent without causing microhemorrhages. Chimeric aducanumab, on the other hand, nudged plaque load down by 20 percent, which was statistically insignificant in this study, and induced microbleeds.
These data suggest ApoE might be a safer target than Aβ itself for reducing plaques, Xiong concluded. Why might this be? Poorly lipidated ApoE4 makes up but a small component of plaques, whereas anti-Aβ antibodies glom on all over them. Xiong suspects the heightened plaque binding by the latter may excessively trigger microglia and astrocytes, leading to neuroinflammation and microhemorrhages. She is currently testing this theory. HAE-4 was licensed to the South San Francisco-based biotech company Denali Therapeutics (see FierceBiotech news; April 2018 news), but is no longer being developed there, according to Joe Lewcock of Denali.
Mechanism of Action. BET proteins bind acetylated lysines (ac) in histone and recruit transcription factors (TF); apabetalone (yellow) competes for this binding site, preventing transcription. [Courtesy of Ewelina Kulikowski.]
An Epigenetic Way to Douse Inflammation
Some therapeutic approaches come from other areas of research, but unexpectedly turn out to have neuroinflammatory effects. Take apabetalone (RVX-208), an investigational small-molecule drug that binds and inhibits bromodomain and extra-terminal (BET) proteins. BET proteins normally recognize acetylated lysines in histones, and then recruit other transcription factors in order to regulate gene expression. Because apabetalone competes for acetylated histone binding within the BET binding region, it prevents BET proteins from fastening to chromatin. Apabetalone was developed by the biotech company Resverlogix, based in Calgary, Canada, and company researchers have reported various treatment benefits for it. According to published papers, it pumps up expression of ApoA-1, the main component of HDL or “good” cholesterol, prevents calcification of blood vessels, and suppresses gene expression pathways associated with kidney disease (McLure et al., 2013; Wasiak et al., 2018; Gilham et al., 2019).
Resverlogix has tested apabetalone for numerous chronic diseases. The furthest advanced is the Phase 3 BETonMACE trial, which enrolls 2,425 high-risk cardiovascular patients with diabetes and low HDL. Apabetalone is also entering a Phase 2a trial of dialysis patients with end-stage kidney disease, and starting a pilot study for pulmonary arterial hypertension.
In these studies, as well as in preclinical mouse work, the researchers noticed that apabetalone calmed peripheral inflammation. Ewelina Kulikowski and colleagues at Resverlogix found that apabetalone turns down expression of complement proteins, part of the innate immune system (Wasiak et al., 2017). This observation led them to test the molecule in cellular and mouse models of neuroinflammation.
In Lisbon, Kulikowski reported that apabetalone inhibits expression of adhesion proteins by endothelial cells, preventing monocytes from sticking to them. This is a crucial step for monocytes to infiltrate into brain (Jul 2018 conference news). In a microglial cell line stimulated with lipopolysaccharide and IFNγ, apabetalone suppressed expression of pro-inflammatory cytokines such as IL-6 and IL-1β, along with complement proteins C3 and C1q. In a wild-type mouse injected intraperitoneally with the inflammatory agent LPS, seven days of treatment with 150 mg/kg apabetalone halved expression of the endothelial inflammation markers E-selectin and ICAM, as well as macrophage and microglial markers CCR2 and CD68. The researchers did not present behavioral data, or experiments in AD mouse models.
Resverlogix researchers wondered if apabetalone treatment could bolster cognition in aging. To test this idea, they decided to include a cognitive substudy in the BETonMACE trial. Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas, an academic consultant to the study, described the experimental paradigm in Lisbon. BETonMACE participants receive 100 mg apabetalone daily for about two years. The primary endpoint is the time until a cardiovascular event such as stroke, heart attack, or death, and the trial continues until 250 such events have occurred. The drug will be deemed a success if it significantly delays adverse cardiac events.
In the cognitive substudy, 467 participants age 70 or older take the Montreal Cognitive Assessment (MoCA) at baseline, every year thereafter, and at study termination. The researchers will compare change from baseline in people on apabetalone and placebo to look for a slowing of cognitive decline. A MoCA of 26 or higher is considered normal cognition, and the average MoCA score at baseline for the whole cohort was 25, Cummings said in Lisbon. Researchers will also analyze subgroups of people who started the trial with mild cognitive impairment; among the 246 people who scored below 26 at baseline, the average MoCA was 22. Topline data from the study are expected this summer.—Madolyn Bowman Rogers
Wasiak S, Tsujikawa LM, Halliday C, Stotz SC, Gilham D, Jahagirdar R, Kalantar-Zadeh K, Robson R, Sweeney M, Johansson JO, Wong NC, Kulikowski E.
Benefit of Apabetalone on Plasma Proteins in Renal Disease.
Kidney Int Rep. 2018 May;3(3):711-721. Epub 2017 Dec 8
PubMed.
Do Immune Cells Promote the Spread of α-Synuclein Pathology?
How does α-synuclein pathology spread? At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, speakers said immune cells bear some of the blame. Markus Britschgi of Roche in Basel, Switzerland, said certain types of inflammation in the intestine modulate α-synuclein accumulation there. In mice, experimental colitis at a young age accelerated α-synuclein pathology in the brain 18 months later, consistent with the idea that misfolded protein can travel from gut to brain. Others implicated brain immune cells in propagation. Seung-Jae Lee of Seoul National University College of Medicine, South Korea, found that mutant α-synuclein oligomers that were incapable of forming fibrils still stimulated aggregation in brain. They appeared to work their mischief by firing up inflammation, suggesting that microglia somehow mediate α-synuclein spread. Together, the findings highlight the role of the immune system in Parkinson’s disease.
First, peripheral immunity. Scientists know that intestinal infections or inflammation can pump up α-synuclein production in the gut, perhaps as part of an antimicrobial defense (Jul 2017 news; Breen et al., 2019; Prigent et al., 2019). This strengthened the idea that Parkinson’s disease might start in the intestine and travel from there to the brain (Jul 2011 news series; Dec 2016 news). People who suffer from inflammatory bowel disorders are at elevated risk of PD, and genetic studies have found shared risk between the two (Jan 2018 news; Apr 2018 news).
While the links are suggestive, no one had yet shown directly that gut inflammation triggered brain pathology. Britschgi and colleagues provoked colitis in 3-month-old transgenic α-synuclein mice by adding dextran sulfate sodium (DSS) to their water. This irritant caused macrophages to invade the lining of the gut wall. In response, enteric neurons lying just below the mucosa, in the submucosal plexus, began to accumulate α-synuclein, although expression of the protein remained unchanged. Britschgi previously reported that such excess α-synuclein persisted for months (Apr 2015 conference news).
Promoting Pathology. More α-synuclein aggregates (brown) accumulate in two brainstem regions of aged transgenic mice that had colitis when young (bottom panels), compared with aged healthy transgenics (top). [Courtesy of Emmanuel Quansah, Patrik Brundin, and Markus Britschgi.]
In Lisbon, Britschgi connected these local effects to brain pathology. In collaboration with Patrik Brundin’s group at the Van Andel Research Institute in Grand Rapids, Michigan, the researchers aged the mice to 12 or 21 months. At 12 months, they saw no difference between the brains of control transgenics and those that had colitis as youngsters. By 21 months, however, the colitis group had six times more α-synuclein aggregates in brainstem regions than controls did (see image at right). These mice had but half as many nigral dopaminergic neurons as controls, suggestive of neurodegeneration.
The finding supports the idea that α-synuclein pathology can propagate from gut to brain. Other studies have shown directly that α-synuclein aggregates injected into the gut travel through the vagus nerve and reach the brain (Holmqvist et al., 2014; Uemura et al., 2018). However, Britschgi said he cannot prove this was the mode of transmission in the Roche study, since they did not cut the vagus nerve to see if that prevented brain pathology.
How do infiltrating immune cells contribute to α-synuclein accumulation in the gut? Previously, Britschgi reported that more α-synuclein built up in mice that lacked fractalkine signaling, a key activator of macrophages and microglia. In Lisbon, he noted that the type of peripheral inflammation also matters. When the researchers irritated the guts of transgenic mice with lipopolysaccharide, α-synuclein did not accumulate, even though macrophages invaded the gut mucosa. The researchers compared the cytokine response in the two types of inflammation, and found that colitis caused IL-6 to spike, while LPS triggered IL-10. Injecting IL-10 into transgenic mice at the same time as DSS dampened the resulting colitis and macrophage infiltration, and prevented α-synuclein buildup. The results suggest that the way macrophages respond to intestinal irritation determines the degree of damage.
Are these mouse data applicable to Parkinson’s? To see if the data were an artifact of α-synuclein overexpression, Britschgi and colleagues repeated the experiments in wild-type mice. As in transgenics, DSS induced α-synuclein deposits in the colon that persisted long after the inflammation had subsided.
Turning to people, the researchers examined intestinal biopsies from 11 people with colitis, 11 with Crohn’s disease, and eight healthy controls. Eight of the colitis patients had macrophages containing α-synuclein in the walls of their guts, and also enteric neurites that stained for α-synuclein accumulation (see image below). Among the Crohn’s patients, four had α-synuclein-positive macrophages in their mucosa but no α-synuclein-positive neurites. Only one of the controls had any α-synuclein reactivity in gut, in a handful of cells. Britschgi noted that they could not tell if the α-synuclein in enteric neurons was aggregated. These data are available online in preprint format (Grathwohl et al., 2019). The data suggest that what macrophages are up to in Parkinson’s pathogenesis should be explored further, Britschgi said.
Synuclein Buildup. α-Synuclein (red) accumulates in enteric neurites (arrowheads) and infiltrating macrophages (arrows) in the intestinal wall of a person with active colitis. [Courtesy of Markus Britschgi.]
Lee made a similar argument, though he approached the question of immune cell involvement from a different angle. Lee is interested in how α-synuclein aggregates propagate within the brain. He noted that when researchers injected aggregated material into mouse brain, it was quickly cleared to undetectable levels. Then, after an incubation period, aggregates appeared and spread through brain. The leading theory holds that this occurs through templated seeding of endogenous α-synuclein by the injected aggregates.
To test this idea, Lee used a mutant form of α-synuclein, V40G, that forms unstructured oligomers but is incapable of forming fibrils. In a test tube, V40G blocks fibrillization of wild-type α-synuclein as well. Thus, this form should prevent templated seeding in vivo, Lee reasoned. The researchers injected either V40G or wild-type α-synuclein into the striata of wild-type mice. To their surprise, V40G seeded aggregates even better than wild-type α-synuclein did. Four weeks after injection, mice that had received V40G had far more α-synuclein pathology in the rhinal cortex than did mice treated with wild-type protein.
Why might this be? The researchers analyzed gene expression in injected brains to glean clues. They found heightened inflammatory and innate immune responses in V40G-treated animals relative to those treated with wild-type α-synuclein. Supporting this, levels of the inflammatory cytokine IL-1β shot up in numerous brain regions after V40G administration, and this spike preceded the spread of α-synuclein aggregates to these regions. Treating mice with the anti-inflammatory drug lenalidomide along with V40G prevented this spike in IL-1β, Lee said. In preliminary experiments, lenalidomide treatment also appeared to ameliorate behavioral deficits in the open field, Y-maze, and wire-hanging tasks; these studies are ongoing.
Based on these findings, Lee proposed a new model of α-synuclein propagation. Perhaps α-synuclein oligomers kick off microglial activation and cytokine release, and this inflammatory microenvironment then aggravates nearby neurons, causing α-synuclein to clump up in their cell bodies. By this logic, rather than α-synuclein aggregates passing directly from neuron to neuron, microglia would be essentially the conveyor belt for α-synuclein pathology.
This new model remains to be tested, but even so, the findings further reinforce the central role inflammation appears to play in neurodegenerative disease, scientists agreed.—Madolyn Bowman Rogers
Forget Fibrils: Lewy Pathology Is More Lipid Than Protein
In Parkinson’s disease, abnormal deposits called Lewy bodies and Lewy neurites pop up in neurons and processes, respectively. In 1998, Maria Grazia Spillantini at the University of Cambridge identified α-synuclein as their major component, providing a key clue to Parkinson’s pathogenesis (Spillantini et al., 1998). Yet despite decades of study, researchers still do not fully understand how these Lewy structures form and what they mean for the health of the cells that carry them. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, two talks from a European research group offered a surprising glimpse at the ultra-substructure of Lewy bodies.
Using cutting-edge microscopy techniques, the researchers found that the center of most Lewy bodies appears to be stuffed with much more lipid than protein. In fact, they say, the deposits represent a mass of undigested membrane fragments, damaged organelles, and other cellular garbage. The researchers also shed new light on what forms of α-synuclein populate Lewy bodies, and how these forms are arranged within neurons. Together, the data offer new clues for deciphering the effect of these structures, as well as preventing formation of Lewy bodies, the speakers suggested. Both studies are currently in preprint form on bioRχiv.
“The two studies complement each other, and paint a sharper picture of Lewy pathology,” co-senior author Markus Britschgi of Roche in Basel, Switzerland, wrote to Alzforum.
Other researchers agreed the data advance the understanding of Lewy pathology. “These findings are exciting and should make us rethink what we have accepted for the past 30 years,” Tiago Outeiro of Göttingen University Medical Center, Germany, wrote to Alzforum (full comment below). Tim Bartels of the U.K. Dementia Research Institute at University College London noted that previous research had linked α-synuclein’s aberrant lipid interactions to cellular toxicity, but not to pathology. “By demonstrating that lipid dysfunction, but not necessarily amyloid formation, is the actual hallmark of the disease, these studies might reconcile past findings and refocus our research,” Bartels wrote (full comment below).
Layered Like an Onion? Antibodies to different forms of α -synuclein reveal concentric spheres of S129-phosphorylated (green), 122-C-terminally-truncated (gold), and 119-C-terminally truncated (red) α-synuclein inside a Lewy body. [Courtesy of Tim Moors.]
Previous research suggested that Lewy bodies form the way amyloid plaques do, with small oligomeric forms of α-synuclein coalescing to form fibrils. According to this theory, these fibrils entrap other proteins and eventually become compacted into a primarily filamentous deposit, the whole process taking up to 10 years. Older microscopic analysis of these bodies revealed a dense, protein-packed core and a peripheral halo of radiating filaments (Arima et al., 1998; Goedert, 2015). In addition to α-synuclein, Lewy bodies contain at least 90 other molecules, including mitochondrial proteins and autophagy-related proteins such as ubiquitin (Wakabayashi et al., 2012). Lewy bodies vary greatly in size, shape, and number between brain regions and donors, however, making them difficult to study.
New microscopic techniques now refine this picture. Academic researchers in Switzerland, the Netherlands, and Germany collaborated with Roche scientists to examine Lewy bodies from postmortem human brain samples. In Lisbon, Amanda Lewis of the University of Basel explained how the group correlated and overlaid scans from light and electron microscopy (EM) in the same block of tissue. This approach, established by Sarah Shahmoradian and led by Henning Stahlberg of the University of Basel, Wilma van de Berg of Amsterdam UMC, and Matthias Lauer and Britschgi at Roche, allowed them to make an unbiased EM survey of all Lewy bodies found by light microscopy. The researchers examined samples taken from the substantia nigra and CA2 of five PD patients. They found 17 Lewy bodies and three Lewy neurites in these samples.
Lewy Flotsam. Electron microscopy unmasks a jumble of lipids, lysosomes (small dark circles), and damaged mitochondria (large circles) at the heart of most Lewy bodies. [Courtesy of Sarah Shahmoradian.]
Contrary to previous research, most of these Lewy bodies appear crammed with lipid structures, rather than proteinaceous filaments. Membrane fragments, vesicles, lysosomes, and misshapen organelles packed together in their cores. Mitochondria appeared frequently, and in some cases formed a shell around the inclusion. Scattered throughout the body were granular proteins, α-synuclein, and patches of disorganized filaments. The α-synuclein was mixed in with membranes and organelles, and did not appear to be in large aggregates.
Lewy neurites had a similar lipid-rich composition. In collaboration with Klaus Gerwert’s group at Ruhr University in Bochum, Germany, the researchers confirmed the presence of lipids using coherent anti-Stokes Raman scattering (CARS), an imaging technique that identifies the chemical composition of substances. Mass spectrometry and Fourier transform infrared (FTIR) spectroscopic imaging provided additional support for the finding.
Only three of the 17 Lewy bodies were stuffed with filaments. Of these, only one had the classic structure, with a protein-packed core surrounded by a filamentous net that traps organelles (Forno and Norville, 1976; Forno, 1996). The scientists are unsure if these were cytoskeletal or α-synuclein filaments (Shahmoradian et al., 2019). The findings raise the question of why previous EM studies had identified mostly filamentous Lewy bodies. It could be because lipid-filled bodies are difficult to distinguish in EM sections; thus, absent direct correlation of scans with light microscope images in this new study, these structures could have been missed. In addition, previous EM tissue preparation techniques tended to be harsh and strip lipids from samples, Britschgi told Alzforum.
The data supports a new theory for Lewy body formation, Lewis said. Perhaps excess α-synuclein disrupts membranes and impairs organelle trafficking, leading these types of debris to pile up. The mess could eventually compact into a Lewy body or Lewy neurite.
Trash Pile. New model of Lewy body formation suggests that α-synuclein wreaks havoc by disrupting membranes, then binds together a loose assortment of cellular garbage. [Courtesy of Sarah Shahmoradian.]
AD/PD attendees were intrigued. Some expressed skepticism, wondering if the data could be an artifact of tissue processing. Lewis said that in vitro controls show their processing methods do not alter tissue composition. In their paper, the authors demonstrate that cytoskeletal filaments and lipid-rich myelin sheaths are unharmed by the same tissue processing,
In the other talk, Tim Moors of Amsterdam UMC added detail on the role of α-synuclein in Lewy body inclusions. Led by van de Berg, Moors’ study combined immunostaining for specific forms of α-synuclein with stimulated emission depletion (STED) microscopy. STED produces an ultra-high resolution image by quenching surrounding fluorophores, leaving the region of interest highlighted. The researchers used antibodies against C-terminally truncated and Ser129 phosphorylated α-synuclein. Both forms are associated with Lewy bodies. Examining postmortem samples from the substantia nigra, hippocampi, and entorhinal cortices of 13 PD patients and six age-matched controls, they discovered a consistent pattern in spatial organization of α-synuclein in midbrain Lewy bodies.
The researchers found that most midbrain Lewy bodies had a layered, onion-like structure. The periphery typically contained pS129 α-synuclein caught in a cage of neurofilament. The dense core was stuffed with aggregated lipids and proteins, including truncated and membrane-associated α-synuclein. Lewy neurites had a similar structure. In the hippocampus and entorhinal cortex, on the other hand, α-synuclein staining appeared uniform throughout most Lewy bodies.
Neurofilament Wheel. A cage-like structure of neurofilament (yellow) encapsulates Lewy bodies. [Courtesy of Tim Moors.]
Do these forms of α-synuclein appear in healthy control brain? Barely any pS129 does, in keeping with this being a purely pathological form of the protein. However, controls did have some 122-truncated α-synuclein that associated with mitochondria, hinting this form could fulfill a physiological function (Moors et al., 2018).
To Moors’ mind, these data suggest that pS129 helps drive Lewy body formation. The researchers proposed that this starts with an excess of α-synuclein in the cytoplasm, which begins to clump with other proteins, membranes, and organelles. The cell may then phosphorylate α-synuclein to prevent fibril formation and trigger degradation of the garbage (Paleologou et al., 2008; Oueslati et al., 2013; Dahmene et al., 2017). When the mass becomes too indigestible, however, the cell encapsulates and compacts it using neurofilament. “α-Synuclein seems to follow its proposed physiological function of binding and organizing membranes, organelles, and vesicles in an orchestrated manner, yet under pathological conditions this seems to go havoc,” Britschgi suggested.
Though the studies focus on different aspects of Lewy bodies, they arrive at similar findings. “The unifying aspect of our two studies is the presence of organellar structures amid enriched different forms of α-synuclein,” Britschgi wrote to Alzforum. He added that the heterogeneity of Lewy pathology necessitates additional research with those new imaging methods. “We hope our findings trigger more studies on Lewy pathology, including mechanistic ones that will help us to identify therapies and biomarkers for Parkinson’s disease and related disorders,” Britschgi wrote.—Madolyn Bowman Rogers
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, Sütterlin R, Huisman E, Ingrassia A, Gier Y, Rozemuller AJ, Wang J, Paepe A, Erny J, Staempfli A, Hoernschemeyer J, Großerüschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, Van IJcken WF, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, Van de Berg WD, Lauer ME.
Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes.
Nat Neurosci. 2019 Jul;22(7):1099-1109. Epub 2019 Jun 24
PubMed.
Keep Your Enthusiasm? Scientists Process Brutal Trial Data
“There is a lot of bad news, but it’s useful bad news.” This line by Randy Bateman of Washington University, St. Louis, captured the mood at the AD/PD conference held March 27 to 31 in Lisbon, Portugal. Whereas in years past scientists filled a paucity of clinical data with educated guesses about what might work, at this meeting they got reams of data to process—all negative as far as the amyloid hypothesis goes. The prevailing attitude was one of learning from mistakes and pleading for patience. Usually after setbacks, the upbeat quotes come out, and so it was here. Some speakers quoted Winston Churchill’s “Success consists of going from failure to failure without loss of enthusiasm.” Others borrowed Benjamin Franklin’s “I have not failed. I’ve just found 10,000 ways that won’t work.”
This spring, scientists found three ways that won’t work. They include trying to slow cognitive decline in symptomatic Alzheimer’s with the anti-Aβ antibodies aducanumab and crenezumab, or with the BACE inhibitor lanabecestat.
On aducanumab, there is no data to report yet. Biogen halted the ENGAGE and EMERGE Phase 3 trials six days before AD/PD (March 2019 news), and in Lisbon did not present; indeed, a scheduled talk was withdrawn. Alas, scientists abide a vacuum no more than nature does, and by the time the 3,892 attendees had been convening for six days, they were trading word in the hallways that the futility analysis had shown not even a hint of efficacy for aducanumab, with placebo and treatment curves superimposed. They speculated why this was either surprising or predictable, but speculation is not data. Sometimes, futility analyses taken during a trial deliver surprises later, once researchers have a chance to dig into the data; this is how Roche resurrected gantenerumab after its SCarlet Road trial stumbled (see below). So by the end of AD/PD, scientists parted on reassurance by Biogen’s Samantha BuddHaeberlein that her company will share Phase 3 data as soon as possible. Soon after, though, any hope that Biogen would proceed with a secondary prevention trial it had been planning were dashed when, on April 24, the company announced an end to the program and removed aducanumab from its pipeline (Biogen Q1 Update).
At AD/PD, attendees challenged pharma researchers. Why in the world would they try more anti-Aβ antibodies or BACE inhibitors when none have worked so far? The researchers defended their deliberative approach of taking time to learn from each other’s failures before scratching a class of drugs, or a target. They noted that the history of drug development is rife with instances where, when multiple companies pursued the same target with slightly different molecules, some stumbled but others learned from those stumbles and later found success. One example is using CETP inhibitors to raise HDL cholesterol, said John Sims of Eli Lilly and Company. Pharma scientists argued it can be difficult to disentangle right away whether a negative trial is due to the molecule, the trial, or the target. Referring to aducanumab’s news, Roche’s Geoffrey Kerchner said, “A press release does not lead the whole industry to stop trials with other molecules in the same class. It is a mistake to pull a drug out of well-designed trials before there is evidence that it does not work.”
Then what is the latest evidence on Aβ antibodies and on BACE inhibitors, the two main ways of targeting amyloid? AD/PD featured detailed data on Roche’s crenezumab and gantenerumab and a bit on Eisai’s BAN2401 (see below). It also featured ample data on Eli Lilly’s lanabecestat and tidbits on two BACE inhibitors currently in the running, Eisai’s elenbecestat and Novartis’ umibecestat (see Part 16 of this series).
Faster With ARIA. In this gantenerumab trial participant, an ARIA-E developed in the same brain region that saw fast and dramatic amyloid reduction. But even in participants who did not develop ARIA, amyloid scans went from positive to negative over the course of two years. [Courtesy of Roche.]
First, crenezumab. Roche announced the end of its two-trial Phase 3 program on January 30 (Jan 2019 news), and at AD/PD it showed the results it has gathered thus far. “Patients are still finishing their follow-up visits, and data are coming in. So the data are preliminary, but today I show what we have because we want to share rapidly,” Susanne Ostrowitzki of Roche/Genentech told the AD/PD audience.
Because this humanized anti-Aβ IgG4 has limited effector function, postulated to result in low risk of ARIA-E, it drew considerable note in the field during the early years of amyloid immunotherapy, before researchers learned that this type of edema is a manageable side effect, not an automatic show-stopper. Crenezumab binds to Aβ monomer and, reportedly at 10-fold higher affinity, to oligomers. It is seen in the periphery of plaques; an area thought to be rich in oligomers; this inspired hope that it might do away with toxic species of Aβ (Jul 2018 conference news).
The two Phase 3 CREAD trials halted this year were evaluating a fourfold higher dose than had been used in Phase 2. Both Phase 2 trials had missed their primary endpoint and, as often happens in this field, post hoc and exploratory data digging hinted at numerically better outcomes in the earlier-stage patients and slower amyloid accumulation at the higher dose. That ray of hope was enough for Roche to go into Phase 3, testing monthly infusions of 60 mg/kg, i.e. three to five grams depending on a participant’s weight.
Two CREAD trials started a year apart; in Lisbon, Ostrowitzki showed data from CREAD 1. By the time of clinical data cut in January, 813 people with prodromal or mild AD had been randomized, 13 percent in both arms had completed the study, 14 percent had discontinued. The crenezumab and placebo groups were well-matched at baseline. At the time the clinical data were cut, each group still had about 300 people in it. Given that this was an interim analysis, the two treatment groups were about 400 strong at baseline and down to the mid-60s by their last, week 105 visit.
The result? Crenezumab did not work at all. On the primary outcome, the CDR-sum of boxes (CDR-SB), the drug and placebo curves sat on top of each other. Ditto for the ADAS-Cog, and for the MMSE. Nothing.
On a functional measure, the ADCS-activities of daily living, the curves looked as if people on crenezumab were doing slightly worse than those on placebo. When the researchers followed up on this by taking the functional aspects of the CDR-SB and broke open the total score into its parts, the differences fell on either side of zero and were inconsistent between timepoints. “The data does not support a drug-placebo difference,” Ostrowitzki said. In other words: It was noise.
Did people whose disease had progressed less respond any better than those who were further along? No. The researchers split the participants two ways: into prodromal versus mild cases as per baseline characteristics, and again by whether they fell below or above 24 on the MMSE alone. They got the same result each time: The cuts did split the trial population as expected, but the crenezumab groups progressed exactly as the placebo groups. “We saw no treatment signal at the earlier versus later stage,” Ostrowitzki said.
Did ApoE make a difference? No. And as an aside, “E4 carrier status had no prognostic effect,” Ostrowitzki said.
At the high dose used in this trial, in this preliminary dataset, crenezumab produced numerically more side effects than placebo but the differences were small. Nothing alarming stood out in the data Ostrowitzki presented.
In toto, crenezumab appears to have reached its target to the extent the researchers expected, but did nothing to treat AD. “Of course this was very disappointing to everyone,” Ostrowitzki said. The full set of CREAD clinical data, and its biomarker data, are not in yet, she added.
A head scratcher? “Crenezumab is quite similar to solanezumab in its binding,” commented Eric Siemers, formerly of Lilly. Siemers was surprised that crenezumab had no efficacy whatsoever, when solanezumab did have a tiny but real benefit at the same stage of AD. Was it affinity? Exposure in the brain? Even at this high dose, most crenezumab would have bound to Aβ in the periphery. Even of the smidgen of crenezumab that entered the brain—Ostrowitzki reported a CSF to serum ratio of 0.21—much would have bound monomer because monomeric Aβ is more abundant in the brain than oligomeric, Siemers noted.
Do You Need ARIA to Clear Amyloid?
With crenezumab biting the dust, scientists asked whether perhaps a clinical benefit cannot be had without plaque removal, and plaque removal cannot be had without ARIA-E, the edema that comes with rapid movement and clearance of amyloid deposits. At AD/PD, Greg Klein of Roche offered this answer: No, ARIA is not necessary. But it does speed things up.
Klein presented data on gantenerumab, Roche’s anti-Aβ IgG1 antibody, which, unlike crenezumab, binds amyloid plaques and strongly activates microglia to clear them. Gantenerumab itself wobbled in 2014, when a futility analysis ended the Phase 3 SCarlet RoAD trial in prodromal AD, but it did not fall. After halting SCarlet RoAD and its fellow Phase 3 trial Marguerite RoAD, Roche decided to convert both into open-label extension studies to gather more data. (Some scientists at AD/PD expressed misgivings about interim futility analyses, which save people from the risk and hassle of negative trials and cut costs, but also deprive scientists of the full set of results.)
In Lisbon, Klein showed results of the gantenerumab open-label extension amyloid PET sub-study. Sixty-nine people had scans at baseline and after one year on gantenerumab; 39 people had three scans, at baseline and after one and two years. Gantenerumab removed amyloid to below the 24 centiloid threshold in all cohorts, no matter whether they started out at 50 or 90 centiloids. “Regardless of baseline amyloid level, or prodromal versus mild subgroup, after two years on gantenerumab, the mean for all groups was below the positivity line,” Klein said.
But is ARIA-E required for this removal? This question can be answered because, while 93 percent of the ARIA-Es in this extension trial were asymptomatic, this type of localized swelling is easily visible on MRI. In Lisbon, Klein showed examples of focal ARIA-E at six months, which resolved by 12 months and, interestingly, occurred in spots where there was a dramatically large amyloid removal as well (see image above). Even so, when Klein compared PET and MRI across all participants, he learned that while ARIA-E marks “hotspots” of rapid amyloid disappearance, amyloid recedes brain-wide even without ARIA. After two years, people who had ARIA-E along the way shed 64 centiloids of amyloid; people without ARIA-E, 56. “It’s a timing issue: Amyloid removal happens fast with ARIA, and more slowly without. ARIA is not required for significant amyloid reduction with gantenerumab,” Klein said.
And no one wants this side effect, even if it is less serious than initially thought. Biogen designed its now-defunct Phase 3 aducanumab trials to minimize ARIA, and concern over this side effect threw a wrench into the 856-patient Phase 2b BAN2401 trial when European regulators ordered sponsors to keep ApoE4 homozygotes off the highest dose.
For its part, Roche again looked to its combined SCarlet and Marguerite RoAD open-label cohort to learn. For example, could they tell who might be most prone to ARIA-E? Looking across time, they saw that a person’s global amyloid load at baseline did not predict; however, regional amyloid offered a hint in that the incidence of ARIA-E was highest in people with a lot of amyloid in the back of their brain. The occipital region is where people with cerebral amyloid angiopathy tend to have the highest burden; CAA accompanies AD and is a risk factor for ARIA-E (Johnson et al., 2007; Gurol et al., 2016).
Start Slow, Go Low
Klein’s colleague Nathalie Pross presented another insight gleaned from the SCarlet/Marguerite RoAD open-label extension cohort. The reason why the near-dead gantenerumab program was resuscitated after the 2014 SCarlet RoAD futility scare was that post hoc analyses suggested there had indeed been a dose-dependent slowing of cognitive decline, albeit only in fast progressors. Also, their CSF tau and neurogranin levels had separated dose-dependently from placebo.
Based on this and other data in the field, Roche decided to up the gantenerumab dose to 1,200 mg. Yet this time they did not rush into another Phase 3, but first explored this new target dose, along with titration schemes to minimize ARIA, in the participants they already had, i.e., the extension cohort, Pross said.
Roche’s in silico Alzheimer’s disease model had predicted that, when given from the start, this dose would cause ARIA-E in 58 percent of participants and that titration might cut this to 25 percent, a rate Roche considers manageable. After trying out titration schemes in the open-label cohort, the actual rate of ARIA across those schemes came in at 28 percent for SCarlet and 32 percent for Marguerite RoAD, Pross reported.
Notably, a person’s ApoE genotype did not affect their risk of ARIA-E during titration to the target dose. Hence Roche is conducting its ongoing Phase 3 GRADUATE trials with the same four-step, nine-month titration in all participants. From month nine until the end of the blinded two-year period, the treatment group will be on 1,020 mg. This slight lowering from the planned 1,200 mg target was enabled by a bioequivalent formulation change along the way, Pross said.
To ease study participants’ travel burdens, they can opt to have gantenerumab or placebo injected under the skin at home for a majority of “visits,” Pross said. The trials include the usual outcome measures, plus exploratory plasma markers as an additional endpoint.
This information is the latest piece of what Roche has learned in a decade of studying gantenerumab, with the trials starting in 2008. Pross said Roche tried to innovate, designing one of the first prodromal trials, first subcutaneous delivery, and embracing the DIAN-TU in 2012. Some participants have been on gantenerumab for nine years, Klein said. When pressed why Roche continues with gantenerumab in light of its prior woes and aducanumab’s downfall, Kerchner said: “We have not seen aducanumab data yet. I do not know why it failed. I do know that with gantenerumab we have put a great deal of work into understanding how dose relates to target engagement and took that knowledge into Phase 3. We have hundreds of patient years of exposure with gantenerumab, and have done our best to learn about the molecule before moving forward.”
Taking note, competing companies are increasingly offering open-label extensions, as well. They see how data collected that way helped Roche better understand their drug; besides, many prospective participants refuse to join a trial unless they get the opportunity to at least receive the drug after the placebo-controlled period is over.
Chad Swanson and colleagues presented at AD/PD that in February 2019, Eisai added a previously unplanned open-label extension to the 856-person Phase 2b study of their anti-Aβ protofibril antibody BAN2401 (Nov 2018 conference news), which ended in summer 2016. Participants from the original, placebo-controlled trial, regardless of whether they completed the 18 months of treatment—and regardless of their ApoE genotypes—are being invited back to receive the highest BAN2401 dose for up to two years total. This means they will come to their clinic dozens more times for biweekly infusions and assessments on top of 42 previous visits the core study already required.
In light of what companies have learned about managing ARIA, people who develop asymptomatic ARIA during this open-label extension can stay on BAN2401 uninterrupted, and people with symptomatic ARIA can go back on the antibody once their ARIA has stabilized or resolved. The researchers hope to learn more about safety and the time course of amyloid removal.
Because health economists agree that monthly antibody infusions won’t become lifelong therapy for millions of Alzheimer’s patients, Eisai scientists are also curious to see what happens after a “drug holiday.” Participants in the long-term extension will have been off BAN2401 for a few years. What did this do to their amyloid, to its downstream biomarkers, and indeed to the response that matters—cognition and function in life? Did the improvements last? And what happens when a person goes back on treatment?
Eisai researchers are hoping to enroll up to 250 people into the open-label extension. It will run until August 2021, alongside the BAN2401 Phase 3 trial, which started in March 2019 to enroll 1,566 people with early symptomatic AD and is set to run until 2024.—Gabrielle Strobel
BACE Inhibitors: Postmortem on One, Live Updates on Two
At the AD/PD 2019 conference held recently in Lisbon, the amyloid hypothesis was reeling from repeated gut punches in the clinic. Both antibodies and BACE inhibitors are facing relentless criticism from academic scientists and biopharma analysts (e.g., May 2019 news). Lost in the cacophony can be the voices of patients and their families urging researchers to push on, and of site leaders who swear they “saw something” among their participants. A little lost as well, amid all the opinion, can be the data itself. Some anti-amyloid investigational antibodies (see Part 15 of this series) and the active vaccine CAD106 are still in Phase 3 or 2/3, as are elenbecestat and umibecestat. In Lisbon, scientists presented and debated data on both these extant BACE inhibitors as well as an extinct one, lanabecestat.
First, the one that doesn’t work, lanabecestat. In June 2018, a futility analysis of its AMARANTH Phase 2/3 trial of 2,219 people with MCI due to AD or mild AD predicted it would fall short of its goal. At the time, Lilly and AstraZeneca stopped not only AMARANTH, which was fully enrolled and 16 months from completion, but also its sister trial DAYBREAK in people with mild AD (June 2018 news). In Lisbon, scientists presented full results on both studies, picking through the wreckage to see what they could learn.
Pierre Tariot of the Banner Alzheimer Institute in Phoenix started off with AMARANTH, the larger of the two. Baseline demographics and clinical assessment showed its groups, gathered at hundreds of sites and dozens of countries around the world, were well balanced. This is a small step forward in the sense that the trials themselves are increasingly poised to pick up a drug benefit once an effective drug comes along. “We know from these presentations that the lanabecestat trials were well done,” said Eric Karran of Abbvie.
Alas, lanabecestat is not that drug, at least not in symptomatic AD. In slide after slide of nearly perfectly superimposed curves, Tariot showed that neither of the two doses tested, 20 or 50 mg per day, moved the primary outcome, or any of the secondary outcomes, even one bit. Lars Lannfelt of Sweden’s Uppsala University captured the mood of many when he said, “Seeing how there was no effect at all was depressing.”
On all of those outcome scales, participants on placebo declined, after both one and two years, as much as one would expect, hence the trial’s negative result can’t be blamed on an aberrant placebo response, Tariot noted. The placebo group’s decline has been a point of contention in trials in years past, so here, too, a small advance was noted. The lanabecestat groups were sizable, boasting more than 700 at baseline and 171–198 who had completed the two-year treatment period when the trial got cut short. In essence, Tariot showed, the trial worked, the drug didn’t.
How safe was lanabecestat? AMARANTH generated a total drug exposure of 1,200 person-years. Lanabecestat was well tolerated, without ARIA, and, most importantly, without the cognitive worsening that had beset Merck’s BACE inhibitor verubecestat and Janssen’s atabecestat. Lanabecestat’s side effects did not warrant stopping, Tariot said. To optimists, this data means that cognitive worsening is not a class effect of BACE inhibition, contrary to previous concerns (Nov 2018 conference news).
That said, the high dose did come with more adverse events and withdrawals. The most common ones considered serious were psychiatric, weight loss of nearly 2 kg, and hair discoloration. Patchy depigmentation of the skin and hair has been reported in mice, rats, rabbits, and dogs for those BACE inhibitors that also block BACE2’s cleavage of pigment cell-specific melanocyte protein (Shimshek et al. 2016; Cebers et al., 2016; Nov 2016 news). Lanabecestat is such a non-selective BACE1/2 inhibitor.
Next up in Lisbon, Lilly’s John Sims presented DAYBREAK data. They matched those of AMARANTH. The data set is smaller because DAYBREAK started after AMARANTH and was still enrolling when both trials ended. Even though DAYBREAK sought only people with mild AD and AMARANTH also enrolled MCI due to AD, their respective populations turned out to be nearly identical, except the former were a tad more impaired on the ADAS-Cog, MMSE, and the RBANS delayed-memory index, Sims showed. And indeed, the parade of “no benefit” slides on the primary and secondary outcomes was nearly the same, as well.
To the ever-hopeful eye, a few of the drug-placebo curves did appear to separate. However, lest anyone read a hint of a treatment benefit into that, Sims noted that the separations were inconsistent across dose and mostly insignificant. He thinks they are noise. “Once you get below 250 people for a longitudinal study, you have a lot of baseline variation bias and survival bias, so you have to be very cautious,” Sims said. When DAYBREAK was halted last summer, it had around 560 people in each group at baseline, but only 18–24 of the earliest enrollees had reached the 78-week time point, so those groups were too small to support much interpretation.
DAYBREAK’s side effect profile was unsurprising, too. The trial did not replicate AMARANTH’s higher dropout rate due to adverse effects on the high dose, but it did replicate the overall pattern of side effects, especially the weight loss and hair discoloration.
Sims and Tariot both emphasized that the full data showed that the futility analysis had made the right call to end this drug program.
What about biomarker data? Lilly presented those, too. Brian Willis and colleagues combined plasma data from both trials and showed that both lanabecestat doses docked blood Aβ40 and 42 levels by 70 to 80 percent. For CSF, the researchers showed AMARANTH data. Aβ40 and 42 levels dropped by 50 to 73 percent, respectively; the alternative cleavage product sAPPα rose and the BACE cleavage metabolite sAPPβ plummeted. In other words, lanabecestat robustly engaged its target. This means the chosen doses tested—and unfortunately disproved—the hypothesis that lanabecestat slows progression of symptomatic AD.
Markers of downstream consequences of target engagement were inconclusive. CSF tau went down but not in a dose-dependent way, making this result hard to interpret, and a substudy of CSF and plasma neurofilament light showed no change with treatment or placebo.
The trials’ substantial stack of brain scans has its own story to tell. In Lisbon, Mark Mintun of Lilly/Avid Radiopharmaceuticals said that the lanabecestat trials had gathered the most imaging data of an Alzheimer’s clinical program so far. It offers lessons not only on what this BACE inhibitor did not do, but also more broadly on how longitudinal biomarkers change relative to disease progression at this early symptomatic stage. Both trials had scheduled florbetapir, flortaucipir, FDG-PET and volumetric MRI scans at two to four time points. Despite the early termination, researchers were able to analyze serial florbetapir scans from 400 participants, serial flortaucipir scans from 354, FDG PET on 298, and MRI on 2,351 participants.
What did they find? First, about the drug. Lanabecestat dose-dependently reduced amyloid, with the high dose drawing out 20 centiloids’ worth of plaques. This result by itself set tongues wagging at AD/PD. There were numerical hints of neurofibrillary tangle reduction, but they fell short of statistical significance. On both amyloid and tau PET, the placebo groups behaved as expected for this disease stage, i.e., florbetapir uptake stayed unchanged and flortaucipir uptake rose. Measured by MRI, the whole brain and the hippocampus area shrank faster in people on lanabecestat than placebo, Mintun showed.
Second, about disease progression. In correlating two-year biomarker change of all groups with change on their clinical outcomes, Mintun tied decreasing FDG PET to worsening across all clinical measures, from ADAS-Cog13 to ADLs to MMSE, FAQ, and CDR-SB. This adds recent, and sizable, multicenter trial data to the existing consensus that FDG PET might serve as an outcome measure in future trials of other drugs. These correlations were not increased by treatment, i.e., unrelated to lanabecestat.
Other biomarker-outcome correlations made less intuitive sense to the audience. For example, Mintun showed that a reduction in florbetapir uptake correlated with worsening on the MMSE. The ADAS-Cog, too, showed a trend toward worse scores with less amyloid. Less amyloid, worse cognition? Several commentators took this to mean that by the time a person has reached the MCI/mild dementia stage of Alzheimer’s, their disease has become independent of amyloid plaques. In other words, the trial confirmed that a person’s amyloid burden tends to level off and then nudge downward as his or her dementia deepens, but the drivers at this stage are factors beyond Aβ, and removing it does not help (Karran et al, 2011).
What about tangles and atrophy for tracking disease progression? In this data set, flortaucipir uptake did not correlate with outcome measures, as was the case for CSF tau. But MRI did. A shrinking brain was bad for people, strongly correlating with worsening across all efficacy measures. As with FDG-PET, this atrophy correlation refers to disease progression itself, not to lanabecestat treatment.
In toto, Mintun showed, two years of high-dose lanabecestat slightly reduced a person’s neuritic plaque load, giving yet more evidence of target engagement. In mildly symptomatic people, this yielded no clinical benefit, and the more distal markers did not respond to lanabecestat treatment. With regard to the field’s broader goal of incrementally validating outcome biomarkers for use in future treatment trials, FDG PET and brain volume both tracked with clinical worsening.
How About the Other BACE Inhibitors?
Given this resounding defeat of lanabecestat in mild AD, do BACE inhibitors have a future in Alzheimer’s treatment? Scientists are not ready to throw in the towel, though compounds that block both BACE1 and 2 equally appear to be a thing of the past. Two compounds currently left standing are Eisai/Biogen’s elenbecestat and Novartis/Amgen’s umibecestat/CNP520. Both claim to be selective for BACE 1 over BACE 2.
Elenbecestat is in the midst of a Phase 3 program called MISSION AD. Its two trials both evaluate 50 mg per day in 1,330 people with early AD, and are expected to read out in 2021. Meanwhile, three posters at AD/PD showed early data on screening tools and tau PET. Eisai’s Michelle Gee and colleagues are evaluating how well a Cogstate test that uses a grocery list to measure immediate and delayed recall predicts brain amyloid positivity among 2,746 people who were screened for participation in MISSION trials. People who scored low on remembering the food items, particularly after a half-hour delay, were more likely to have brain amyloid deposition than those who aced the list. Similar data were shown for a brief Cogstate battery of cognitive tests. This kind of evaluation helps trial sponsors find tools to cut back the number of expensive amyloid scans during screening.
Also at AD/PD, Andrew Stephens and colleagues at Life Molecular Imaging, previously Piramal, showed preliminary results of a tau PET substudy in the MISSION AD trials. Among 45 amyloid-positive participants scanned thus far with LMI’s tau tracer PI-2620, 27 had a brain-wide positive scan. Of those, 13 had overt neocortical tangles outside the mesial-temporal cortex; 14 had subtler deposition only in their hippocampal region, or diffuse or focal uptake. Eighteen participants had a negative PI-2620 scan. While the study is still enrolling, this early data suggest that the amyloid-positive, mildly symptomatic MISSION AD participants span a range of tau pathology as they are starting on elenbecestat.
Among AD researchers, BACE1 selectivity has become a touchpoint for justifying effort on a new generation of inhibitors. What about it for elenbecestat and umibecestat?
Elenbecestat is said to preferentially block BACE1 over BACE2 (Nov 2018 conference news). According to Teiji Kimura of Eisai, elenbecestat is not specific for BACE1; however, the dose used in Eisai’s clinical trials, which was guided in part by the 40 percent reduced Aβ production seen for the protective APP mutation, might allow for a preferential effect on BACE1. In a presentation made to investors and media on April 23, Eisai reported that elenbecestat binds BACE1 with an affinity of 19 nanomolar and BACE2, 67 nM, amounting to 3.53-fold selectivity. Pharma companies frequently synthesize their competitors’ compounds to compare them with their own, and this same Eisai presentation posts lanabecestat’s selectivity as being twofold, and verubecestat’s as being 0.17-fold, i.e., as preferring BACE2 over BACE1.
According to Eisai, depigmentation cropped up neither in animal studies nor clinic trials of elenbecestat thus far. At its most recent, eighth, meeting, MISSION’s data-safety-monitoring board reviewed safety and cognition data gathered thus far and recommended that the trials continue.
Of the BACE inhibitors currently in clinical trials, Novartis/Amgen’s umibecestat has a peer-reviewed publication to show it prefers BACE1 over BACE2 threefold. Moreover, it is present in skin only at low concentrations, meaning it has little chance to encounter targets that would lead to depigmentation (Neumann et al., 2018; Dobrowolska Zakaria and Vassar 2018). In Lisbon, Derya Shimshek of Novartis told Alzforum that Novartis, too, has seen no hair discoloration in preclinical studies. This side effect is not dangerous but nettlesome because it breaks the blind.
The API GENERATION program comprises two global Phase 2/3 secondary prevention trials of umibecestat/CNP520 in people whose risk of developing AD symptoms is high because they carry the APOE4 allele and are between 60 and 75 years old. Both studies are enrolling, and so far, there has been no statement of cognitive harm, according to Tariot. By design, one GENERATION study includes people without elevated brain amyloid. In other words, this trial, as well as the API ADAD crenezumab trial in Colombia, will be the first to generate data on how anti-amyloid treatment affects people with preclinical AD, at an earlier stage than all other trials.
Meanwhile, Novartis researchers at AD/PD in Lisbon presented animal data suggesting their compound may not cause the hippocampal mossy fiber damage scientists believe might partly account for cognitive worsening. Novartis’ Carine Kolly and colleagues picked up recent work by Robert Vassar’s group at Northwestern University in Chicago, which had described this phenotype in adult conditional BACE knockout mice. Their hippocampal mossy fibers were shorter and disorganized, just as in knockouts of the axon guidance gene, and BACE1 substrate, CHL1 (Sep 2018 news).
Kolly and colleagues first confirmed that Novartis’ in-house BACE1 knockout mice had this phenotype. Then they examined brain tissue from a long-term rat toxicology study to see if CNP520 produced it, too. The rats had received a six-month course of up to 200 mg/kg/day of umibecestat, a 10-fold excess over the highest dose used in people. This slashed Aβ levels yet left the length and organization of mossy fibers in the hippocampus intact.
Why would CNP520 not inhibit CHL1 processing? Kolly noted that cells harbor two pools of BACE1, one in the trans-Golgi network and one in early endosomes. CHL1 is cleaved in the trans-Golgi; APP in endosomes. While the genetic knockouts lack BACE1 in both pools, CNP520 preferentially accumulates in endosomes. This subcellular localization, Kolly hypothesized, may explain why the drug leaves CHL1 alone.
CHL1 is but one of about 45 known BACE substrates, and sorting out what BACE inhibition does to them is important. It is a complicated task, because changes in the processing of some of those substrates might mask a cognitive benefit, besides causing outright side effects. “We need more specific inhibitors that avoid other substrates. Ideally, you want to block only BACE1’s interaction with APP,” Lannfelt told Alzforum.
Separately at AD/PD, Shimshek and colleagues at Novartis showed preclinical data hinting that umibecestat could potentially treat cerebral amyloid angiopathy (CAA), a common pathological feature of Alzheimer’s and by itself a cause of stroke and dementia. APP23 mice who ate umibecestat with their chow for six months deposited less amyloid in their cerebral small blood vessels. The effect was dose-dependent.
What Next?
Opinion at AD/PD about the future of BACE inhibition diverged widely. While some scientists believe the approach should be scrapped, others insist that lower doses ought to be tried at earlier disease stages in a preventive mode.
“Lanabecestat’s toxicity was not bad for a terminal disease. In that way, I see Alzheimer’s like cancer. We now know BACE inhibition reduces amyloid, by 20 percent as per PET. We have to find a way to test these drugs earlier,” said an undeterred Colin Masters of the University of Melbourne, Australia. He and others envision future trials that enroll people whose amyloid deposition just crosses the PET threshold, push it below the threshold with an Aβ antibody, and then keep it there with a low dose of a BACE1 inhibitor. Vassar, who cloned BACE but also described several side effects of knocking it out, remains hopeful, too. “Let’s not give up on BACE inhibitors. We still do not know when to treat and how much to inhibit. Every failure teaches us something,” Vassar said.
On May 10, Eisai announced that the Alzheimer’s Clinical Trials Consortium (Dec 2017 news) chose elenbecestat and BAN2401 for primary and secondary prevention trials starting up early next year (company press release).—Gabrielle Strobel



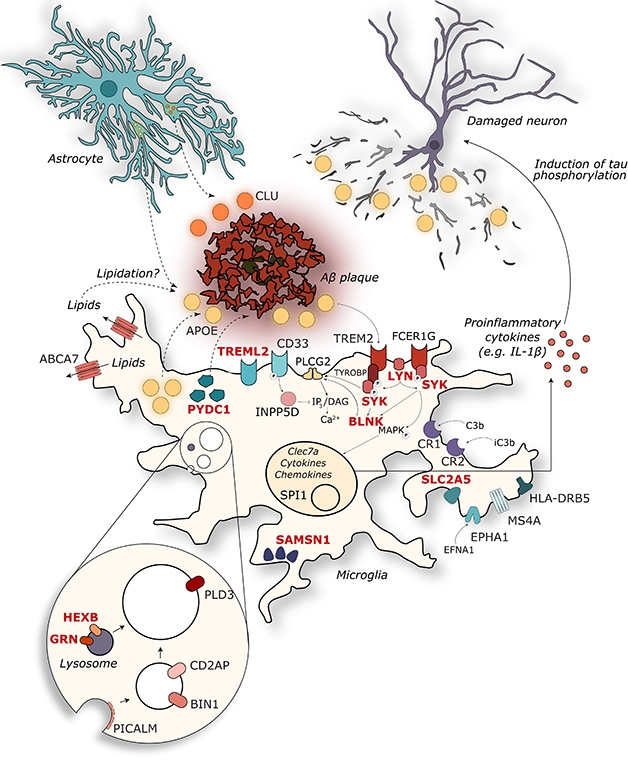
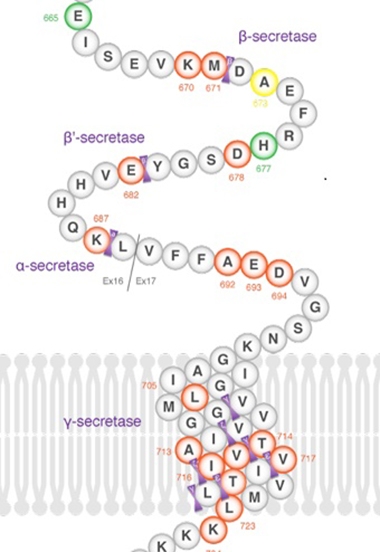






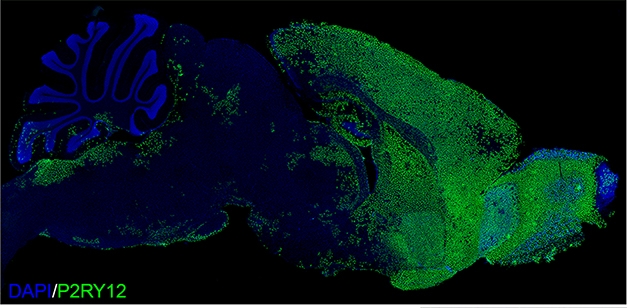
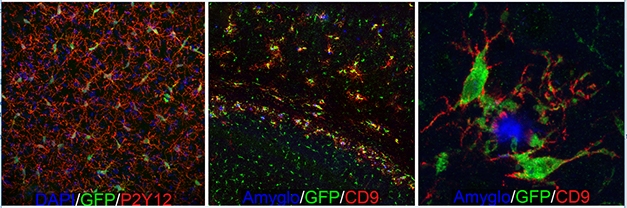
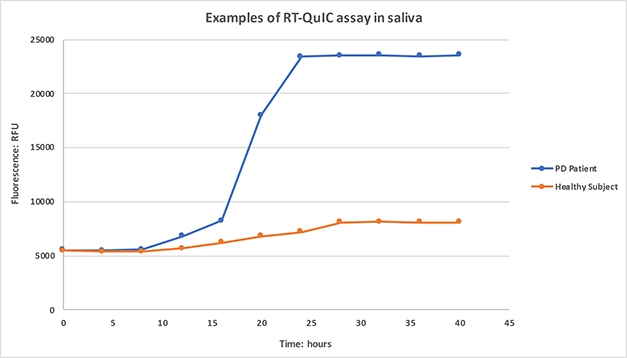
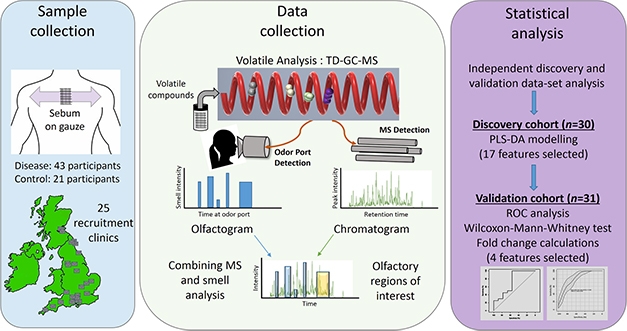










Comments
No Available Comments
Make a Comment
To make a comment you must login or register.