Gene Links Childhood Neurodegeneration to Mitophagy
Quick Links
Neurodegenerative disease almost always strikes in adults, but some extremely rare forms can crop up in adolescents or even children. Genetic clues to abnormal pathways that underlie these early onset disorders might help scientists better understand late-onset diseases such as Alzheimer’s. A study in the September 1 American Journal of Human Genetics reports that rare loss-of-function mutations affecting both copies of a gene called SQSTM1/p62 lead to a form of juvenile neurodegeneration characterized by gait abnormalities and ataxia. Heterozygous variants in this same gene have been linked to a number of other neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Since p62 takes part in recycling worn-out mitochondria (aka mitophagy), the findings emphasize how crucial this process is for neural health.
“This is an interesting paper that reports a third neuromuscular/neurodegenerative phenotype for SQSTM1,” wrote Teepu Siddique, Northwestern University, Chicago, to Alzforum. Siddique had previously reported that SQSTM1 mutations cause ALS, while others subsequently linked variants in the gene to FTD (Fecto et al., 2011; Rubino et al., 2012). He noted that the involvement of lower motor neurons in some affected children in this study, and upper motor neurons in another, strengthen the link between p62 and ALS. Heterozygous mutations in SQSTM1, which is short for sequestosome 1, also have been linked to Paget disease of the bone (Rea et al., 2014). Valosin-containing protein, another FTD/ALS gene, has been linked to Paget’s, as well.
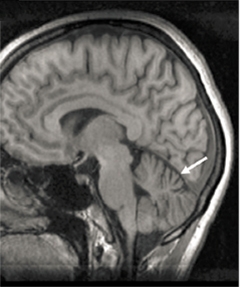
Shrinking cerebellum.
MRI unveils cerebellar atrophy (arrow) in a 31-year-old patient with neurodegenerative disease that began in childhood. [Courtesy of Haack et al., 2016. AJHG.]
Mitochondrial proteins, including those that help turn over and replenish healthy organelles, have been implicated in a number of neurodegenerative disorders, such as Parkinson’s disease and ALS (Heo et al., 2015). When mitochondria get old, autophagy adaptor proteins such as OPTN and SQSTM1 help dispose of the organelles by tagging them for autophagy (Geisler et al., 2010).
Haack and Klopstock came across SQSTM1/p62 when they were looking through patient exomes for mutations that might explain childhood neurodegenerative disorders. From four unrelated families they studied nine people who had trouble with speech and posture, including limited control of their upper bodies. These symptoms had slowly worsened since the patients were 7 to 15 years old. Most also had some combination of dystonia, uncoordinated eye movements, and cognitive decline. MRIs revealed cerebellar atrophy in four of them (see image above). Several patients had been followed for decades and were in their mid-40s when last examined. Because the pattern of inheritance hinted at a recessive disorder, the researchers searched for biallelic gene disruptions.
It turned out that each patient had two dysfunctional copies of the SQSTM1 gene. Three affected siblings born to healthy parents of German ancestry had a homozygous c.2T>A variant in exon 1, predicted to cause loss of protein function. In another family, three sisters harbored homozygous deletions of nucleotides 311 and 312 that resulted in a frameshift variant and premature stop codon 48 amino acids later (p.Glu104Valfs48*) that segregated with disease. These girls were born to parents from the United Arab Emirates who didn’t realize they were related before they got married. Two more patients from a Finnish family and one of Kurdish descent carried a c.286C>T mutation in both copies of the gene that led to a premature stop codon (p.Arg96*). This variant appears to have arisen independently in both families. No family members who were heterozygous for any of these mutations had symptoms. Furthermore, out of 127,000 control exomes in two separate databases, no one had mutations that affected both copies of SQSTM1, suggesting that such changes are relatively rare.
How did these mutations affect the gene? The researchers found no p62 at all in fibroblasts from one patient with the c.2T>A variant in exon 1 or in three patients with the c.286C>T (p.Arg96*) mutation. Probing for SQSTM1 mRNA from two fibroblasts with the latter mutation revealed drastically reduced transcripts in primary fibroblasts. When depolarized by the chemical CCCP, damaged mitochondria in these cells congregated less around the nucleus than did those organelles in normal fibroblasts. A previous study suggested that SQSTM1 is required for this clustering, which appears to be a prerequisite for mitophagy (Narendra et al., 2010). In addition, fewer autophagosomes formed in affected fibroblasts. “These observations suggest that in dividing cells, SQSTM1/p62 contributes to the early regulation of mitophagy,” wrote the authors. “The present study establishes absence of SQSTM1/p62 as a molecular defect underlying a childhood- or adolescence-onset neurodegenerative disorder,” they added.
That patients live upwards of 40 years with no SQSTM1—at least in fibroblasts—and develop a limited neurological phenotype that suggests cells can compensate to some degree, perhaps with redundant proteins and pathways, the authors proposed. The study broadens the set of diseases associated with the SQSTM1 protein and supports the role for impaired autophagy in neurodegenerative disease, they wrote.
Zhenyu Yue at the Icahn School of Medicine at Mount Sinai in New York pointed out that SQSTM1/p62 also plays an important role in the autophagy of ubiquitinated protein aggregates, and disrupting that could contribute to the underlying defect in this form of childhood neurodegeneration. However, he was surprised that the absence of SQSTM1/p62 causes such a severe phenotype in people, since knocking out the gene in mice has a more subtle non-motor effect marked by accumulation of hyperphosphorylated tau and neurodegeneration (Ramesh Babu et al., 2008).—Gwyneth Dickey Zakaib
References
Paper Citations
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011 Nov;68(11):1440-6. PubMed.
- Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St George-Hyslop PH, Scarpini E, Gallone S, Pinessi L, TODEM Study Group. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012 Oct 9;79(15):1556-62. Epub 2012 Sep 12 PubMed.
- Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations--bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014 Jul 1;325(1):27-37. Epub 2014 Jan 30 PubMed.
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015 Oct 1;60(1):7-20. Epub 2015 Sep 10 PubMed.
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010 Feb;12(2):119-31. Epub 2010 Jan 24 PubMed.
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010 Nov;6(8):1090-106. PubMed.
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008 Jul;106(1):107-20. PubMed.
Further Reading
Papers
- Morisada N, Tsuneishi S, Taguchi K, Yagi R, Nishiyama M, Toyoshima D, Nakagawa T, Takeshima Y, Takada S, Iijima K. [A woman with beta-propeller protein-associated neurodegeneration identified by the WDR45 mutation presenting as Rett-like syndrome in childhood]. No To Hattatsu. 2016 May;48(3):209-12. PubMed.
Primary Papers
- Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P, Maffezzini C, Lönnqvist T, Suomalainen A, Gorza M, Kremer LS, Graf E, Hartig M, Berutti R, Paucar M, Svenningsson P, Stranneheim H, Brandberg G, Wedell A, Kurian MA, Hayflick SA, Venco P, Tiranti V, Strom TM, Dichgans M, Horvath R, Holinski-Feder E, Freyer C, Meitinger T, Prokisch H, Senderek J, Wredenberg A, Carroll CJ, Klopstock T. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am J Hum Genet. 2016 Sep 1;99(3):735-43. Epub 2016 Aug 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.