TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
Quick Links
When the going gets tough, microglia rely on TREM2 to get going. That was the common conclusion from two papers that investigated the role of the myeloid receptor in mouse models of Alzheimer’s disease and stroke. One study, led by Marco Colonna at Washington University in St. Louis, reported that TREM2 promotes microglial survival and helps the cells clear plaques. The other, led by Midori Yenari at the University of California, San Francisco, found that TREM2 supports microglial cleanup of damaged tissue following stroke, which improves neurological recovery. Both groups also discovered potential TREM2 ligands—lipids and nucleic acids—that could trigger the receptor during AD and stroke, respectively. Overall, TREM2 emerged as a broad sensor of cellular damage that may provide crucial reinforcement for the brain’s macrophages when they’re needed most.
TREM2 took the neurodegenerative disease field by storm in 2012, when researchers discovered that a variant of the receptor triples AD risk (see Nov 2012 news). It has also been linked to frontotemporal dementia, Parkinson's, and amyotrophic lateral sclerosis (see Feb 2014 news). People completely lacking the receptor develop Nasu-Hakola disease, a lethal form of neurodegeneration accompanied with bone abnormalities. TREM2 is expressed on myeloid cells, including microglia in the brain, and reportedly promotes anti-inflammatory, phagocytic responses in those cells (see Hickman and El Khoury, 2014). The identification of this immune receptor as a risk factor for neurodegenerative disease underscores neuroinflammation’s central role there (see Heneka et al., 2015), and researchers have been racing to understand how TREM2 fits in.
Colonna, who originally cloned the TREM2 gene, sought to explore how the receptor affected microglial function in a mouse model of AD. As reported at conferences and in previous papers, Colonna and other researchers had found that plaque burden either stayed the same or shrank in TREM2-deficient mice crossed with APP/PS1 mice (see Jun 2014 news and Dec 2014 conference coverage). For the current study, Colonna crossed TREM2 KO mice to 5xFAD mice instead, as this AD model develops plaques more slowly. He reported some of his findings, now also published on February 26 in Cell, at a recent Keystone meeting covered by Alzforum (see Feb 2015 conference coverage).
In summary, first author Yaming Wang and colleagues found that 5xFAD mice lacking TREM2 had a heavier plaque burden in the hippocampus than 5xFAD mice expressing the receptor. Microglia failed to gather around plaques or switch on activation genes in TREM2-deficient mice, and many of the cells that remained were apoptotic. The researchers determined that TREM2 signaling synergized with that from colony-stimulating factor receptor 1 (CSFR1), thus providing a survival signal that was necessary in situations where CSF levels were low. Colonna hypothesized that this would occur in the AD brain, where microglia proliferate and suck up copious amounts of CSF.
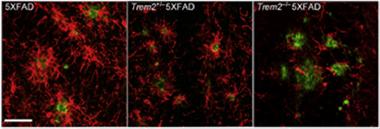
Crucial Reinforcement.
Activated Iba1+ macrophages (red) crowd around amyloid plaques (X34, green) in 5xFAD mice (left). The cells mount less of a response in mice with one copy of TREM2 (center), and hardly budge in mice lacking both (right). [Image courtesy of Wang et al., Cell 2015.]
What turned on TREM2 survival signaling during this response? Colonna hypothesized that TREM2 may respond to anionic lipids, which are found in cell membranes, because previous reports suggested that the receptor recognized anionic macromolecules from bacteria, such as lipopolysaccharide and peptidoglygan, and cell-associated lipids (see Daws et al., 2003, and Cannon et al., 2012). The researchers confirmed that this was the case: TREM2 signaling switched on in response to phospholipids, which are exposed on apoptotic cells, and sphingolipids, which are released by damaged myelin. Interestingly, many of the lipids failed to activate TREM2 containing the AD-associated R47H mutation, suggesting the receptor may not switch on in people who carry this genetic variant. Overall, Colonna’s study suggested that in response to lipids, TREM2 provides a microglial survival signal that encourages the cells to clean any nearby debris.

TREM2 Rallies the Troops.
In response to lipids, TREM2 supports microglial survival, promoting the cells to clean up plaques and neuronal debris. [Image courtesy of Wang et al., Cell 2015.]
John Hardy of University College London, commented that Colonna's work added another layer of complexity to TREM2’s role in AD. “When we found loss of function TREM2 mutations associated with Alzheimer's disease, we had assumed that it disrupted the interaction between Aβ and the microglia,” he wrote. “This work confirms that this view, while generally accurate, is an oversimplification and that the interaction is with the lipids around the plaque rather than with Aβ itself.”
Michael Heneka of the German Center for Neurodegenerative Diseases in Bonn commented that the study was the most in-depth analysis of microglial TREM2 function in an AD model to date. He was intrigued by the finding that lipids could trigger TREM2 and promote microglial survival. “TREM2 expression at Aβ plaque sites can be interpreted as an attempt to survive the local inflammatory and toxic milieu, a prerequisite to restrict Aβ accumulation by phagocytosis or release of degrading proteases,” Heneka wrote.
Yenari’s findings, published February 25 in the Journal of Neuroscience, dovetailed with Colonna’s. First author Masahito Kawabori and colleagues investigated the role of TREM2 in a middle cerebral artery occlusion model of stroke—a far more drastic and acute setting of cell death than AD. Immediately following the stroke, both normal and TREM2-deficient mice performed poorly on tests of neurological function. For example, they walked in circles and missed rungs as they scrambled across a horizontal ladder. However, while normal mice started to regain some of these skills after only one day and performed at nearly pre-stroke levels on the ladder task after two weeks, TREM2-deficient mice recovered more slowly and never got close to their pre-stroke performance levels.
Two weeks after the stroke, the researchers found that while about 70 percent of the damaged tissue had been cleared from the brains of normal mice, nearly 90 percent of it remained in TREM2-deficient brains. It is possible that the clean-up failure could have prevented synaptic repairing and rebuilding of damaged axons, which could account for the animals’ dismal recovery, Yenari told Alzforum.
What prevented the TREM2-deficient animals from cleaning up the damage? The researchers found that sub-par microglial recruitment and poor phagocytosis were likely to blame. Around the damaged area in normal mice, they observed an influx of the microglia-expressing activation markers IB4 and CD68. They also noted a striking abundance of “foamy macrophages,” which stained positive with Oil Red O due to their consumption of lipids, presumably derived from dead cells and debris. However, activated cells were present in far fewer numbers in TREM2-deficient mice, and foamy macrophages were virtually absent. Three-dimensional confocal imaging revealed that nearly 40 percent of the activated macrophages in normal mice were in direct contact with dying cells near the stroke site, whereas less than 4 percent of cells in TREM2-deficient mice made contact. All in all, the findings suggested that TREM2 expression on microglia facilitated the clearance of damaged tissue following stroke.
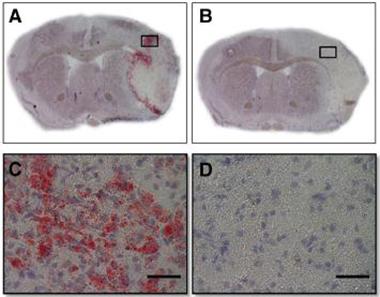
TREM2 Draws a Foam Bath.
Chock-full of lipids from damaged cells, foamy macrophages (red) clean up the mess at the infarct site following stroke (A and C). The cells are absent in TREM2-deficient mice (B and D). [Image courtesy of Kawabori et al., Journal of Neuroscience, 2015.]
What triggered TREM2 during stroke? First, the researchers tested whether TREM2 and its mystery ligand were in the same place at the same time. They found that TREM2 expression ramped up around the infarct site, where it co-localized with CD11b+ macrophages and peaked in expression one week after stroke. In parallel experiments, the researchers stained the brains of mice with a TREM2-Fc fusion protein, which should bind to any potential TREM2 ligands. Expression of these ligands paralleled that of TREM2 itself, except the ligand co-localized with dying neurons near the infarct site.
Yenari had a hunch that nucleic acids could activate TREM2. After all, nucleic acids are known to bind other immune receptors, and spill out of necrotic neurons following stroke. To test this, the researchers used their TREM2 fusion protein as bait in a chromatin immunoprecipitation assay on whole-brain lysates from mice after stroke. Indeed, the TREM2 protein associated with nucleic acids in the lysate. The researchers next used a TREM2-expressing reporter cell line, which turns on GFP when TREM2 is triggered, to confirm that nucleic acids could stimulate the receptor. They detected GFP in response to purified nucleic acids, or to conditioned media from dying neurons that had been starved of oxygen and glucose.
Colonna commented that while both his and Yenari’s identification of TREM2 ligands will need to be confirmed with further assays, the results fit well together. It would make sense that TREM2 could bind both anionic lipids as well as nucleic acids, which are polyanions, Colonna said. Yenari agreed, and commented that nucleic acids may play a more important role in activating TREM2 during stroke, when bursting necrotic cells dump out nucleic acid entrails. In AD, in which cells tend to die more slowly, lipids could be the predominant TREM2 ligand.
Both Colonna and Yenari acknowledged that infiltrating macrophages, in addition to resident microglia, could contribute to plaque or debris clearance in their models. Neither study fully attempted to distinguish between the cells. However, for one experiment, Colonna crossed APP/PS1 mice with CX3CR1-GFP mice. In these mice, microglia, but not infiltrating macrophages, express GFP in the brain. In this scenario, Colonna reported that TREM2-deficiency resulted in a marked decrease in CX3CR1+ cells around plaques, suggesting that resident microglia were predominantly affected by loss of TREM2. However, neither researcher can rule out a contribution from infiltrating macrophages.
Both Yenari and Colonna agreed that the function of the cells and their role in protecting the brain are far more important than their origin.—Jessica Shugart
References
Alzpedia Citations
News Citations
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- TREM2 Variant Doubles the Risk of ALS
- TREM2 Mystery: Altered Microglia, No Effect on Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- United in Confusion: TREM2 Puzzles Researchers in Taos
Research Models Citations
Paper Citations
- Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol. 2014 Apr 15;88(4):495-8. Epub 2013 Dec 16 PubMed.
- Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015 Mar;16(3):229-36. PubMed.
- Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol. 2003 Jul 15;171(2):594-9. PubMed.
- Cannon JP, O'Driscoll M, Litman GW. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics. 2012 Jan;64(1):39-47. Epub 2011 Jul 29 PubMed.
Further Reading
Papers
- Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, Otero K, Piccio L. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015 Mar;129(3):429-47. Epub 2015 Jan 29 PubMed.
Primary Papers
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015 Mar 12;160(6):1061-71. Epub 2015 Feb 26 PubMed.
- Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, Nakamura MC, Yenari MA. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. 2015 Feb 25;35(8):3384-96. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Klinik und Poliklinik für Neurologie
Neuroinflammatory mechanisms have emerged as an important feature and pathogenic event in Alzheimer’s disease. Recently, mutations in TREM2 have been identified as risk factors for the development of the sporadic type of this neurodegenerative disease. In this study, the Colonna group publishes the first profound analysis of microglial TREM2 function in murine AD models. One of the most intriguing findings of this study is that TREM2-deficient microglia seem to show an impaired reaction to Aβ deposition. Since TREM2-deficient 5xFAD mice showed an increase in the overall Aβ load in the hippocampus, this suggest that, at the investigated time point, TREM2 mediated mechanisms that restrict the deposition of Aβ. In keeping with this, TREM2 deficiency impaired microglial recruitment to the site of Aβ deposition. Importantly, the authors excluded, at least by in vitro experiments, that TREM2 deficiency affects microglia Aβ phagocytosis or degradation directly.
Instead TREM2 seems to be involved in microglial survival mechanisms and TREM2 deficiency increased microglial apoptosis, possibly linked to restricted colony-stimulating factor 1 levels. Alternatively, TREM2-deficient cells may harm themselves by an increased release of TNFα, although several types of microglial cell death need to be considered (Kim and Li , 2013; Jung et al. 2005). Thus, TREM2-deficient microglia seem to not survive the Aβ challenge and therefore fail to mount an appropriate clearance response, in line with previous findings showing that improving microglial phagocytosis in vivo can restrict Aβ deposition (Heneka et al., 2013).
Another important finding of this study is that TREM2 is not activated by Aβ itself, as previously suggested, but by certain anionic membrane phospholipids, a a response that was severely limited by the human R47H mutation, which has been linked to sporadic AD. Therefore, TREM2 expression at Aβ plaque sites (Frank et al., 2008; Lue et al., 2014) can be interpreted as an attempt to survive the local inflammatory and toxic milieu, a prerequisite to restrict Aβ accumulation by phagocytosis or release of degrading proteases.
Overall this study further highlights the role of microglia in neurodegeneration and in particular in Alzheimer’s disease. Similar to previous studies, (e.g., Bradshaw et al., 2013) it points to microglial uptake and degradation of Aβ as an important method for restricting the peptide's accumulation. Given the plethora of GWAS-identified mutations that are potentially linked to immune function (Lambert et al., 2013), it can be expected that further disease-relevant microglial functions will be discovered.
Naturally, these findings fuel the hope of developing therapeutics that modify microglia functions. For this to be successful, we need to consider not only the different innate immune mechanisms, but the precise disease stage when they manifest.
References:
Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013 Jul 11;4:e716. PubMed.
Jung DY, Lee H, Suk K. Pro-apoptotic activity of N-myc in activation-induced cell death of microglia. J Neurochem. 2005 Jul;94(1):249-56. PubMed.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674-8. Epub 2012 Dec 19 PubMed.
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008 Oct;56(13):1438-47. PubMed.
Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, Walker DG. TREM2 Protein Expression Changes Correlate with Alzheimer's Disease Neurodegenerative Pathologies in Post-Mortem Temporal Cortices. Brain Pathol. 2014 Sep 3; PubMed.
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, , Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013 Jul;16(7):848-50. PubMed.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossù P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer's Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer's Disease, Alzheimer's Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O'Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P, Wang J, Uitterlinden AG, Rivadeneira F, Koudstgaal PJ, Longstreth WT Jr, Becker JT, Kuller LH, Lumley T, Rice K, Garcia M, Aspelund T, Marksteiner JJ, Dal-Bianco P, Töglhofer AM, Freudenberger P, Ransmayr G, Benke T, Toeglhofer AM, Bressler J, Breteler MM, Fornage M, Hernández I, Rosende Roca M, Ana Mauleón M, Alegrat M, Ramírez-Lorca R, González-Perez A, Chapman J, Stretton A, Morgan A, Kehoe PG, Medway C, Lord J, Turton J, Hooper NM, Vardy E, Warren JD, Schott JM, Uphill J, Ryan N, Rossor M, Ben-Shlomo Y, Makrina D, Gkatzima O, Lupton M, Koutroumani M, Avramidou D, Germanou A, Jessen F, Riedel-Heller S, Dichgans M, Heun R, Kölsch H, Schürmann B, Herold C, Lacour A, Drichel D, Hoffman P, Kornhuber J, Gu W, Feulner T, van den Bussche H, Lawlor B, Lynch A, Mann D, Smith AD, Warden D, Wilcock G, Heuser I, Wiltgang J, Frölich L, Hüll M, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Jöckel KH, Klopp N, Wichmann HE, Dickson DW, Graff-Radford NR, Ma L, Bisceglio G, Fisher E, Warner N, Pickering-Brown S. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013 Dec;45(12):1452-8. Epub 2013 Oct 27 PubMed.
View all comments by Michael HenekaIcahn School of Medicine at Mount Sinai
The results of these two papers are consistent with a model of AD etiology that can be derived from the network analysis of AD (Rosenthal and Kamboh, 2014; Zhang et al., 2013) and TREM2 (Forabosco et al., 2013). This model strongly implicates dysregulation of efferocytosis (i.e., apoptotic cell clearance or, more generally, defective clearance of cellular "debris") in the etiology of AD.
For example, the three major "pathways" previously shown to be enriched in GWA studies for LOAD (lipid/sterol efflux, innate immune cell function, and endocytosis) (Jones et al., 2010) are key components of efferocytosis (Ravichandran and Lorenz, 2007; Poon et al., 2014; A-González and Castrillo, 2010). Moreover, network analysis of human genetic variants associated with LOAD (Rosenthal and Kamboh, 2014) or of human brain gene co-expression data associated with TREM2 (Forabosco et al., 2013) also points to efferocytosis. This scavenging function of microglia and macrophages is critical to inflammation resolution and tissue repair after infection and injury. However, it is also known to play an important role in the maintenance of tissue homeostasis (Davies et al., 2013).
A better understanding of 1) the role of efferocytosis/clearance of cellular “debris” in the maintenance of brain tissue (including myelin and synapses) and 2) the gene network that supports this biological process (which is likely to include APOE, TREM2, TREML2, ABCA7 [an orthologue of the C. elegans efferocytosis gene ced-7], ABCA1, MEGF10, ABCG1, ELMO1, SORL1/retromer, C1Q, LXR, RXR, TRIP4, and several other candidate AD loci/genes) (Hsieh et al., 2009; Takahashi et al., 2005; de Freitas et al., 2012; Jehle et al., 2006; Hamon et al., 2006; Yvan-Charvet et al., 2010; Cash et al., 2012; Kiss et al., 2006; A-Gonzalez et al., 2009; Ruiz et al., 2014) could shed some light on the mystery of AD etiology beyond the amyloid hypothesis (Seong and Matzinger, 2004; Medzhitov 2008; Kotas and Medzhitov, 2015) and inspire the development of novel therapeutic approaches for this devastating disease (Schadt et al., 2009).
References:
Rosenthal SL, Kamboh MI. Late-Onset Alzheimer's Disease Genes and the Potentially Implicated Pathways. Curr Genet Med Rep. 2014;2:85-101. Epub 2014 Mar 22 PubMed.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 Apr 25;153(3):707-20. PubMed.
Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, Weale ME, Ryten M. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 Dec;34(12):2699-714. PubMed.
Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, Gerrish A, Pahwa JS, Jones N, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, Heun R, Kölsch H, van den Bussche H, Heuser I, Peters O, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Rüther E, Carrasquillo MM, Pankratz VS, Younkin SG, Hardy J, O'Donovan MC, Owen MJ, Williams J. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One. 2010;5(11):e13950. PubMed.
Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007 Dec;7(12):964-74. PubMed.
Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014 Mar;14(3):166-80. Epub 2014 Jan 31 PubMed.
A-González N, Castrillo A. Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim Biophys Acta. 2011 Aug;1812(8):982-94. Epub 2010 Dec 28 PubMed.
Rosenthal SL, Kamboh MI. Late-Onset Alzheimer's Disease Genes and the Potentially Implicated Pathways. Curr Genet Med Rep. 2014;2:85-101. Epub 2014 Mar 22 PubMed.
Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, Weale ME, Ryten M. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 Dec;34(12):2699-714. PubMed.
Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013 Oct;14(10):986-95. Epub 2013 Sep 18 PubMed.
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009 May;109(4):1144-56. Epub 2009 Mar 19 PubMed.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005 Feb 21;201(4):647-57. PubMed.
de Freitas A, Banerjee S, Xie N, Cui H, Davis KI, Friggeri A, Fu M, Abraham E, Liu G. Identification of TLT2 as an engulfment receptor for apoptotic cells. J Immunol. 2012 Jun 15;188(12):6381-8. Epub 2012 May 9 PubMed.
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006 Aug 14;174(4):547-56. PubMed.
Hamon Y, Trompier D, Ma Z, Venegas V, Pophillat M, Mignotte V, Zhou Z, Chimini G. Cooperation between engulfment receptors: the case of ABCA1 and MEGF10. PLoS One. 2006 Dec 27;1:e120. PubMed.
Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010 Jun 25;106(12):1861-9. Epub 2010 Apr 29 PubMed.
Cash JG, Kuhel DG, Basford JE, Jaeschke A, Chatterjee TK, Weintraub NL, Hui DY. Apolipoprotein e4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem. 2012 Aug 10;287(33):27876-84. PubMed.
Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006 Nov 21;16(22):2252-8. PubMed.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009 Aug 21;31(2):245-58. Epub 2009 Jul 30 PubMed.
Ruiz A, Heilmann S, Becker T, Hernández I, Wagner H, Thelen M, Mauleón A, Rosende-Roca M, Bellenguez C, Bis JC, Harold D, Gerrish A, Sims R, Sotolongo-Grau O, Espinosa A, Alegret M, Arrieta JL, Lacour A, Leber M, Becker J, Lafuente A, Ruiz S, Vargas L, Rodríguez O, Ortega G, Dominguez MA, IGAP, Mayeux R, Haines JL, Pericak-Vance MA, Farrer LA, Schellenberg GD, Chouraki V, Launer LJ, van Duijn C, Seshadri S, Antúnez C, Breteler MM, Serrano-Ríos M, Jessen F, Tárraga L, Nöthen MM, Maier W, Boada M, Ramírez A. Follow-up of loci from the International Genomics of Alzheimer's Disease Project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry. 2014 Feb 4;4:e358. PubMed.
Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004 Jun;4(6):469-78. PubMed.
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008 Jul 24;454(7203):428-35. PubMed.
Kotas ME, Medzhitov R. Homeostasis, Inflammation, and Disease Susceptibility. Cell. 2015 Feb 26;160(5):816-827. PubMed.
Schadt EE, Friend SH, Shaywitz DA. A network view of disease and compound screening. Nat Rev Drug Discov. 2009 Apr;8(4):286-95. PubMed.
Case Western Reserve University
The intriguing finding of the Wang et al. study is that lipids “activate” wild-type TREM2 and turn on NFAT signaling pathways whereas the R47H variant of TREM2, which is a risk allele for AD, PD, FTD, and ALS, is almost completely inactive in the NFAT reporter assay.
NFAT singling regulates expression of pro-inflammatory cytokines TNF-α, IL-2, IFNg, etc. Thus, the charged lipids—presumably released from apoptotic neurons and coating the amyloid plaques—should activate WT microglia and stimulate the release of inflammatory cytokines, whereas those expressing R47H-TREM2 should not. By implication, WT-TREM2 should be proinflammatory and R47H-TREM2 should not promote inflammation in response to apoptotic cells. This seems to run counterintuitive to the common finding that increased inflammation is observed in the brains of patients with all four neurodegenerative diseases indicated above, and there is increasing evidence that chronic neuroinflammation is toxic to the brain function and initiates neurodegeneration.
One thing to keep in mind is that the NFAT reporter assay was performed by overexpressing WT-TREM2 or R47H-TREM2 in 2B4 reporter T-cells. TREM2 has an extremely short cytoplasmic tail and is known to signal by binding another membrane protein ,DAP12/TYRO-BP, which possesses a longer cytoplasmic tail with an immunoreceptor tyrosine-based activation (ITAM) motif. From the information in the manuscript, it seems that Wang et al. transfected TREM2 alone and not TREM2+DAP12. Also unclear is whether 2B4 T-cells express endogenous DAP12 and if so, what the stoichiometry of overexpressed TREM2 to endogenous DAP12 is. Thus, at present it remains an open question whether the effects of R47H mutation on NFAT signaling reported here shed any light on the role of TREM2 in AD pathogenesis or are due to overexpression of the protein in a non-microglial cell line. Future studies will need to resolve the conundrum of how R47H-TREM2, which does not seem to promote inflammation, increases the risk for neurodegeneration.
View all comments by Sanjay PimplikarMake a Comment
To make a comment you must login or register.