Will Next-Gen BACE Inhibitors Dodge Side Effects?
Quick Links
While BACE inhibitor trials are unfolding, more than 100 experts in the field gathered for the 2nd Kloster Seeon Meeting on BACE Proteases in Health and Disease, held September 25-27 near Munich. Researchers are encouraged that no major side effects have yet reared their ugly heads, but even so, safety worries still inspire active research toward a new round of drugs. In Seeon, researchers described new assays to spot off-target effects, new insight into why fading hair color is the most common problem seen in BACE knockout mice, and new inhibitors that selectively target BACE1 over BACE2. “I think we are on the verge of seeing the next generation of BACE inhibitors,” said Robert Vassar of Northwestern University, Chicago, who co-organized the meeting. “I was not sure this was possible, but now it seems to be happening.”
With BACE, a Picky Drug is a Good Thing
Like any other drug, BACE inhibitors can have both on-target and off-target side effects. Scientists believe the latter led to the eye toxicity that stopped clinical trials of the first BACE inhibitors (see Mar 2011 conference news). More specifically, the assumption has been that by also blocking cathepsin D, another aspartyl protease, BACE1 inhibitors prevented the normal turnover of photoreceptors in the eye, causing lipofuscinosis. In Seeon, Douglas Johnson from Pfizer, Cambridge, Massachusetts, put any lingering doubt about this scenario to rest.
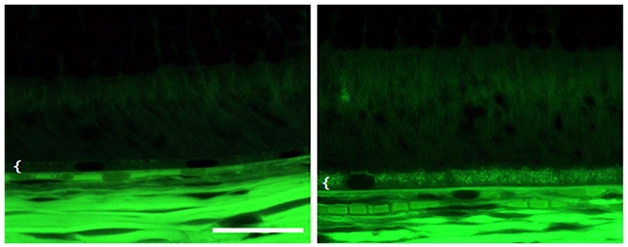
Eye Tox. Autofluorescenece microscopy of rat retina shows the BACE inhibitor PF-9283 (right) disrupts the retinal pigment epithelium (white bracket). [Image courtesy of Douglas Johnson, Nature Communications.]
Johnson used photoaffinity versions of BACE inhibitors to covalently label whatever they bind to in retinal epithelial cells. Then, in an unbiased fashion, he identified those targets by mass spectrometry (see Zuhl et al., 2016). He found that PF-9283, a BACE inhibitor Pfizer developed, showed high affinity for cathepsin D. In competition experiments, LY2811376, one of Eli Lilly’s early BACE inhibitors, and CP-108671, a renin protease inhibitor, both prevented PF-9283 from binding cathepsin D, showing that these compounds also had high off-target affinity for the protease. In contrast, LY2886721 did not, confirming reports that this compound is much more selective for BACE than other aspartyl proteases.
That confirmation aside, Johnson cautioned that test-tube assays for cathepsin D inhibition should be taken with a grain of salt. He found that the affinity of PF-9283 for cathepsin D was two orders of magnitude higher in cells than in vitro. Stefan Lichtenthaler, German Center for Neurodegenerative Diseases in Munich, thought this was astounding. Furthermore, in an exposure-response analysis, in which Johnson correlated IC50s of various compounds with eye epithelium damage in rats, only the cellular IC50s predicted toxicity. “These are very important revelations for drug development,” said Lichtenthaler. “This suggests that scientists who design new inhibitors need to run cell-based assays to determine selectivity,” he told Alzforum. While it remains to be proven why some inhibitors have higher affinity for cathepsin D in cells, it may come down to the compound’s accumulation in acidic environments, since this protease works mostly in the lysosome.
Eye toxicity aside, scientists continue to worry about on-target side effects as well. Importantly, all BACE inhibitors currently in trials block both BACE1 and BACE2. While the latter seems to have fewer substrates and is less active in the brain than BACE1, it may still underlie some unwanted effects, such as the fur discoloration seen in mice. Scientists are also concerned that long-term use of inhibitors might bring to light new BACE2-related effects. Could they develop BACE1-selective compounds to take a cleaner shot at the target?
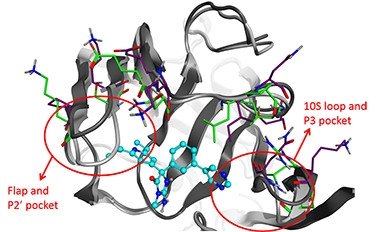
BACE Details.
Overlapping X-ray crystallographic structures of BACE1 (dark gray, blue side chains) and BACE2 (light gray, green side chains) reveal differences (red circles) at the active site that researchers might exploit to develop BACE1-specific inhibitors (turquoise and blue). [Image courtesy of Dieder Moechars.]
Dieder Moechars and colleagues at Janssen Research and Development, Beerse, Belgium, have done just that. Moechars’ team took advantage of the known crystal structures of BACE1 and 2 to design drugs that bind the former but not so much the latter. The researchers noted that because BACE2 has a more rigid active site, it might restrict compounds that the BACE1 cleft would allow in (see image at left). He illustrated this by comparing co-crystal structures of BACE1 and BACE2 together with an inhibitor developed by Wyeth that has about 500-fold higher affinity for BACE1 (see Malamas et al., 2010). Janssen researchers have now improved substantially on that, developing compounds topping out at 7,000-fold higher affinity for BACE1. Janssen plans to select one of these for clinical development early next year, Moechars said.
“This will help allay concerns about hypopigmentation or blocking cleavage of BACE2 substrates in the pancreas,” said Vassar. “Even though BACE2 knockouts are fairly normal, we need to be concerned about blocking BACE2,” he said. Vassar also noted that these new compounds are more potent inhibitors of BACE1, which would translate to lower doses in the clinic. “That means much lower potential for off-target side effects,” he said. “These compounds seem to have lots of advantages.”

Rabbit of a Different Color. Dutch belted rabbits treated for four months with verubecestat (right) lose pigment in their coats. [Image courtesy of Lynn Hyde, Matthew Kennedy, Merck Research Labs.]
What about selectivity against BACE1 substrates other than APP? That idea may not be so far-fetched. After all, γ-secretase modulators can shift cleavage of the APP C-terminal fragment (CTF) while allowing the secretase to process Notch, though these compounds, too, have run into toxicity problems at the preclinical stage. Moechars said Janssen scientists are searching for an APP-selective inhibitor. They want a compound that binds to APP and prevents it from coming into contact with BACE1. With this in mind, they devised an assay for compounds that block production of CTF from APP even while allowing processing of neuregulin1. The researchers had to screen 1.2 million compounds to turn up one that fit the bill. “It proves the concept, but we have to use micromolar amounts, making it far from an ideal inhibitor,” said Moechars. Researchers were intrigued, but wondered how efficient this approach would be in practice. Since the substrate has to be blocked, not the catalyst, some questioned whether enough drug can be delivered into the brain to do the job.
As for the inhibitors currently in trials, most of them seem to mimic the loss of coat color seen in BACE knockouts (see image above). How might that manifest in people? If scientists knew, no one at Seeon was willing to say out loud. Lynn Hyde from Merck, Kenilworth, New Jersey, noted that reports of skin or hair color change in BACE inhibitor trials are scant. Since the trials are still blinded, no firm conclusions can be drawn, though on the face of it such effects would seem to be pretty obvious to participants and trial staff.
To begin to probe this question, Hyde has looked for underlying causes of color loss in mice. She examined changes in gene expression in the skin during anagen phase, the period during which hair grows from follicles and becomes pigmented. Hyde compared gene expression in control animals with that in mice given the BACE inhibitor MBI-3. The drug suppressed expression of 18 genes, including BACE2 and proteins known to be involved in pigmentation such as PMEL17 and tyrosinase. In the BACE1/2 double knockouts generated at Merck, Hyde found the same 18 genes to be suppressed. All of them lie downstream of microphthalmia-associated transcription factor, a major regulator of pigmentation. Hyde said this gene expression signature might serve as a biomarker correlating with BACE inhibitor effects on pigmentation. She does not know if BACE processes MiTF, but said that would be worth exploring.
Tracking BACE Inhibition
One way to mitigate side effects of BACE inhibition would be to find a dose range that suppresses Aβ accumulation while allowing sufficient processing of other substrates. Lichtenthaler reported on a proteomics approach to monitor substrate cleavage by measuring levels of their shed ectodomains in the CSF. Researchers at the meeting thought this might be a good way to test if cleavage of substrates other than APP is being spared (see Part 3 of this series).
For APP itself, researchers have long used Aβ levels in the CSF to gauge efficiency of BACE inhibition; however, Mathias Jucker, University of Tubingen, Germany, reported that this data may be difficult to interpret (see Schelle et al., 2016). APP/PS1 mice, for example, accumulate robust amyloid plaques in the brain by 7.5 months of age, while their CSF Aβ falls about threefold. In contrast, when 1.5-month-old animals ate food pellets laced with the BACE inhibitor NB-360, they accumulated no plaques in the brain over the next six months, but their CSF Aβ fell just the same. Because both plaque formation and BACE inhibition reduce the level of CSF Aβ, the latter may be a misleading marker of BACE activity, said Jucker. This holds in a treatment paradigm as well, he said. If begun when APP23 mice are 15 months old and have rampant plaques, 6.5 months’ worth of treatment with NB-360 reduced CSF Aβ levels, but CSF Aβ also fell in untreated animals.
Jucker proposed a different marker to track BACE processing of APP, and it is none other than tau. Previously, Stephan Käser in Jucker's lab reported that tau climbs in the CSF of transgenice APP/PS mouse models of AD as Aβ plaques begin to accumulate. This happens even though the animals show no tau pathology or neuronal loss (see Jul 2013 news). Would CSF tau respond to BACE inhibition?
Jucker and colleagues developed a highly sensitive, single-molecule array immunoassay (Simoa) for tau that can measure the protein in CSF of young, normal mice and in transgenic animals before they develop overt pathology. He used it to test the effect of NB-360. At Seeon, Jucker reported that in both the APPPS1 prevention paradigm and in the APP23 treatment case, the BACE inhibitor lowered CSF tau while CSF tau climbed in untreated animals. “CSF tau could be a very valuable marker to predict the efficacy of BACE inhibitors in clinical trials,” Jucker said.
He further hinted that another up-and-coming marker of neurodegeneration, neurofilament light chain (NfL), responds to BACE inhibitors, as well. That could be even more valuable, since Jucker has reported that blood NfL tracks disease progression (see Jun 2016 news).
While researches wondered why tau and NfL are released into the CSF in the absence of overt neurodegeneration, they were cautiously optimistic about these data, saying they represent a real breakthrough. “We don’t have a blood biomarker for any neurodegenerative disease yet,” noted Vassar, adding, “These data are extremely interesting and potentially very important for the field.”—Tom Fagan
References
News Citations
- Barcelona: Out of Left Field—Hit to The Eye Kills BACE Inhibitor
- BACE Inhibition and the Synapse—Insights from Seeon
- Cerebrospinal Fluid Tau Climbs in Aβ Mouse Models
- Blood NfL Looks Good as Progression and Outcome Marker
Therapeutics Citations
Research Models Citations
Paper Citations
- Zuhl AM, Nolan CE, Brodney MA, Niessen S, Atchison K, Houle C, Karanian DA, Ambroise C, Brulet JW, Beck EM, Doran SD, O'Neill BT, Am Ende CW, Chang C, Geoghegan KF, West GM, Judkins JC, Hou X, Riddell DR, Johnson DS. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of β-secretase inhibitors. Nat Commun. 2016 Oct 11;7:13042. PubMed.
- Malamas MS, Barnes K, Johnson M, Hui Y, Zhou P, Turner J, Hu Y, Wagner E, Fan K, Chopra R, Olland A, Bard J, Pangalos M, Reinhart P, Robichaud AJ. Di-substituted pyridinyl aminohydantoins as potent and highly selective human beta-secretase (BACE1) inhibitors. Bioorg Med Chem. 2010 Jan 15;18(2):630-9. Epub 2009 Dec 6 PubMed.
- Schelle J, Häsler LM, Göpfert JC, Joos TO, Vanderstichele H, Stoops E, Mandelkow EM, Neumann U, Shimshek DR, Staufenbiel M, Jucker M, Kaeser SA. Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimers Dement. 2016 Oct 14; PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.