Could Disposing of Damaged Mitochondria Treat Alzheimer’s Disease?
Quick Links
Malfunctioning mitochondria accumulate in the Alzheimer’s disease brain. Could they be purged? In the February 11 Nature Neuroscience, researchers led by Vilhelm Bohr at the National Institute on Aging, Baltimore, argue that these defective organelles play a key role in disease progression. In both animal and cellular models, revving up their disposal lessened the hallmark pathologies of AD: amyloid-β plaques and phosphorylated tau. In addition, learning and memory behavior bounced back to normal in treated worms and mice. “We believe mitophagy is a central process early in Alzheimer’s disease, and could be a therapeutic target,” Bohr told Alzforum.
- Mitophagy is compromised in AD brain.
- Stimulating it reduces Aβ/tau pathology and improves memory in animal models.
- Phosphorylated tau prevents mitophagy by sequestering parkin.
“This is a remarkable and outstanding paper,” Flint Beal at Weill Cornell Medical College, New York, wrote to Alzforum. “It ties tau phosphorylation and amyloid pathology to mitochondrial dysfunction and defective mitophagy. Aging, the most important risk factor for AD, is also linked to reductions in mitophagy.”
The relationship between mitochondrial damage and AD pathology seems to run both ways, with each worsening the other (Nov 2009 news). Recently, researchers led by Jürgen Götz at the University of Queensland in Brisbane, Australia, elucidated one mechanism by which tau pathology contributes to mitochondrial mismanagement. They report in the February 1 EMBO Journal that cytoplasmic tau binds to the ubiquitin ligase parkin, preventing parkin from reaching damaged mitochondria and triggering their disposal via mitophagy.
“Together with other studies, these papers show clear evidence of a nexus between mitochondrial dysfunction and protein aggregation,” said Russell Swerdlow at the University of Kansas in Kansas City. It remains unclear which phenomenon occurs first. Swerdlow speculated the order might vary in different forms of the disease, with familial AD more likely triggered by protein aggregation, and sporadic AD by aging, dysfunctional mitochondria (Aug 2013 news).
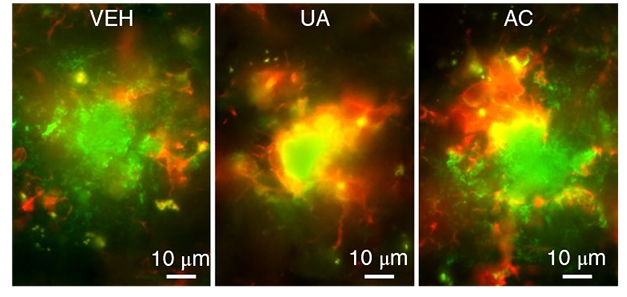
Mitochondria Aid Phagocytosis. In AD mice treated with a mitophagy enhancer (center and right), more microglia (red) feast on plaques (green) than in untreated AD mice (left). [Courtesy of Fang et al., Nature Neuroscience.]
Previous research has established that neurons in AD brain contain fewer healthy, intact mitochondria and more that are broken, oversized, and misplaced (May 2001 news; Jul 2009 news). Cells maintain their pool of mitochondria mainly via lysosomal degradation of damaged organelles. The kinase PINK1 tags defective mitochondria and recruits parkin, which in turn ubiquitinates mitochondrial proteins, flagging the organelle for disposal in lysosomes. Mutations in either of these genes cause Parkinson’s disease, but have not been linked to AD (Apr 2004 news; Apr 2014 news; Sep 2015 news).
Bohr and colleagues wondered what role mitophagy might play in AD progression. Joint first authors Evandro Fang, Yujun Hou, and Konstantinos Palikaras measured the number of mitochondria associated with lysosomes in postmortem AD hippocampus, finding about half as much mitophagy as in control brain. They found the same thing in human neurons generated from iPS cells from AD patients. One of these lines carried an APP mutation, and a second had two copies of ApoE4. In both, proteins that initiate mitophagy, TBK1 and ULK1, were half as active as in control neurons. TBK1 variants cause familial forms of amyotrophic lateral sclerosis and frontotemporal dementia (Feb 2015 news; Mar 2015 news).
Would boosting mitophagy ameliorate AD? The authors first screened for compounds that enhanced mitophagy in the roundworm Caenorhabditis elegans. They identified an antibiotic, actinonin, and a plant compound, urolithin A. They fed urolithin A to a worm that expresses human Aβ42, and found it lowered levels of the peptide throughout the body. Notably, these worms treated with either urolithin A or actinonin learned to avoid a noxious chemical as quickly as wild-types did. Treatment likewise restored normal memory to a worm model of tau pathology (Fatouros et al., 2012). In both models, the improvement depended on mitophagy, as it did not occur in PINK1 mutants.
Turning to mice, the authors fed urolithin A or actinonin to APP/PS1 animals for two months. Mitophagy in hippocampal neurons ramped up to normal, while the number of damaged mitochondria fell to control levels. Amyloid plaque burden in the hippocampus fell by two-thirds, and treated animals performed as well as wild-types in the Morris water and Y mazes.
How does mitophagy lower amyloid? Perhaps via the innate immune system. In treated animals, the authors detected more activated microglia surrounding and engulfing plaques than in controls (see image above). Microglia in these mice were more phagocytic than those in untreated mice, as measured by cell shape and protein expression. In addition, these microglia boasted healthy mitochondria similar to those in wild-type mice, while untreated APP/PS1 microglia accumulated threefold more damaged mitochondria than controls did. Possibly, phagocytosis becomes impaired in untreated APP/PS1 mice because this process requires a great deal of cellular energy, and thus needs healthy mitochondria, the authors speculated.
Swerdlow suggested another possibility. He pointed to recent studies that indicate aggregated Aβ and tau can end up inside mitochondria, where they are either cleared by mitochondria themselves, or eliminated when mitophagy grinds up the organelles (Sorrentino et al., 2017; Du et al., 2017). Both Sorrentino et al. and Du et al. found that enhancing mitophagy lowered amyloid aggregation, in agreement with Bohr’s data. “Mitochondria are like trash bags, and mitophagy takes out the trash,” Swerdlow suggested.
Moreover, Shirley ShiDu Yan and colleagues at the University of Kansas in Lawrence reported that stimulating mitophagy with PINK1 improved synaptic plasticity and memory in AD mice. “The present study from Fang et al. supports our discovery,” Yan wrote to Alzforum.
What about tau? Because APP/PS1 mice develop little tau pathology, Bohr and colleagues turned to 3xTg AD mice, which carry a tau mutation on top of APP and PS1 mutations, and form tangles. Treating them with urolithin A for one month inhibited tau phosphorylation and restored memory in the Y maze and object recognition tests. Intriguingly, previous research from Eva Mandelkow at the German Center for Neurodegenerative Diseases in Bonn found that a tau kinase, MARK2, also regulates PINK1 and mitochondrial transport (Matenia et al., 2012). Bohr noted that in AD, tau pathology associates more closely with cognitive decline than amyloid does, suggesting that targeting tau pathology by boosting mitophagy could help patients.
Bohr and colleagues are testing this in a clinical trial run by Steen Hasselbalch at Copenhagen University Hospital, Denmark. AD patients will take nicotinamide riboside, a Vitamin B3 variant that boosts nicotinamide adenine dinucleotide (NAD+). NAD+ precursors are known to stimulate mitophagy. Bohr said an advantage of this dietary supplement is that it has few side effects. NAD+ precursors are beginning to be evaluated for neurodegenerative and other conditions (Feb 2019 news).
Besides testing nicotinamide riboside in people, Bohr plans to further dissect how tau and mitochondria interact in worms. Prior studies have blamed abnormal tau for instigating mitochondrial dysfunction (Manczak and Reddy, 2012; Duboff et al., 2013; Eckert et al., 2014).
Götz and colleagues added to this literature by showing that cytosolic tau blocks mitophagy. First author Nadia Cummins found that overexpressing either human wild-type or P301L tau in mouse neuroblastoma cells prevented initiation of mitophagy after mitochondria had sustained damage. She traced the cause to a lack of parkin recruitment to these organelles and determined by co-immunoprecipitation that both types of tau directly bound parkin. Testing tau fragments, she found that the amino-terminal end of tau was responsible. Notably, tau did not interact with parkin that was already attached to mitochondria. In short, tau appears to sequester parkin in the cytosol, preventing it from reaching these organelles, the authors concluded. In AD, hyperphosphorylated tau dissociates from microtubules and builds up in cytoplasm.
Bohr noted that the findings dovetail with his. The interaction of tau and parkin strengthens the evidence that mitochondrial dysfunction may crop up early in AD, he said.—Madolyn Bowman Rogers
References
News Citations
- New Triple Transgenic Shows Mitochondrial Damage by Tau, Aβ
- Studies Suggest Mitochondria Changes Precede Aging, Alzheimer’s
- Mitochondrial Damage in Alzheimer's Disease
- Mitochondrial Break-up: Alzheimer’s Alters Fusion, Fission
- Pink Mutations Link Parkinson’s Disease to Mitochondria
- Novel Ubiquitin Modification Ties Two Risk Genes for Parkinsonism
- PINK1 Can Act Alone to Destroy Mitochondria, But Parkin Helps
- TANK-Binding Kinase 1 Rumbles in as New ALS Gene
- Second Study Salutes TANK-Binding Kinase 1 as ALS Gene
- In Small Trial, EH301 Appears to Halt Progression of ALS
Research Models Citations
Paper Citations
- Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet. 2012 Aug 15;21(16):3587-603. PubMed.
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017 Dec 14;552(7684):187-193. Epub 2017 Dec 6 PubMed.
- Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain. 2017 Dec 1;140(12):3233-3251. PubMed.
- Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM. Microtubule Affinity-regulating Kinase 2 (MARK2) Turns on Phosphatase and Tensin Homolog (PTEN)-induced Kinase 1 (PINK1) at Thr-313, a Mutation Site in Parkinson Disease: EFFECTS ON MITOCHONDRIAL TRANSPORT. J Biol Chem. 2012 Mar 9;287(11):8174-86. PubMed.
- Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012 Jun 1;21(11):2538-47. PubMed.
- Duboff B, Feany M, Götz J. Why size matters - balancing mitochondrial dynamics in Alzheimer's disease. Trends Neurosci. 2013 Jun;36(6):325-35. PubMed.
- Eckert A, Nisbet R, Grimm A, Götz J. March separate, strike together--role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. 2014 Aug;1842(8):1258-66. Epub 2013 Sep 17 PubMed.
Further Reading
News
- Protein Screen Links Mitochondrial Regulator to Alzheimer’s Disease
- So Immature—Aβ Stymies Mitochondrial Protein Processing
- A BAD Mitochondrial Dehydrogenase—A Good AD Drug Target?
- Do Mothers’ Mitochondria Magnify Dementia Risk?
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- NO Kidding? Mitochondria Fission Protein Linked to Neurodegeneration
- Aβ and Mitochondria—When It Reigns, They Pore
- Aging and Aβ Hit Mitochondria Function
Primary Papers
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019 Mar;22(3):401-412. Epub 2019 Feb 11 PubMed.
- Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Götz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019 Feb 1;38(3) Epub 2018 Dec 11 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Case Western Reserve University
Case Western Reserve Universit
This is an outstanding study. It not only adds significant evidence supporting a critical role of mitochondrial dysfunction in the pathogenesis of Alzheimer’s disease, but also specifically suggests mitophagy as a promising therapeutic target for AD.
Different from the previous neuron-centric study of mitochondrial (dys)function in AD, it is most interesting and significant that this study demonstrated that mitophagy is also impaired in the microglia in AD mouse brain, and that mitophagy stimulation enhances the phagocytic efficiency of microglia to clear amyloid plaques along with mitigation of NLRP3/caspase-1-dependent neuroinflammation.
While the authors suggested that enhanced phagocytic efficiency in microglia is likely due to the restored supply of required energy by improved mitochondrial homeostasis, detailed mechanisms warrant further investigation. Overall, the study demonstrated the neuroprotective and restorative potential of mitophagy enhancement in AD.
However, since mitochondrial mass is reduced in pyramidal neurons in AD (Hirai et al., 2001), and current evidence supports the notion of impaired mitochondrial biogenesis in AD (Qin et al., 2009; Sheng et al., 2012), the pursuit of mitophagy enhancers alone as a therapeutic may be limited.
References:
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001 May 1;21(9):3017-23. PubMed.
Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009 Mar;66(3):352-61. PubMed.
Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J Neurochem. 2012 Feb;120(3):419-29. PubMed.
University College London
Institute of Neurology, University College London
This paper from Fang et al. represents an impressive amount of work examining the interplay between dysfunctional mitophagy, inflammation, and AD pathologies in multiple disease models. These data add to multiple studies implicating dysfunctional mitophagy in PD (reviewed in (reviewed in McWilliams and Muqit, 2017), a growing number of papers demonstrating a number of FTD/ALS genes are involved in the mitophagy process, such as TBK1, SQSTM, OPTN and VCP (Karbowski and Youle, 2011; Heo et al., 2015; Kim et al., 2013), and a few recent studies suggesting a role for mitophagy in AD (Sorrentino, et al., 2017; Du et al., 2017; Checler et al., 2017; Martín-Maestro et al., 2016). Thus, mitophagy is emerging as a crucial mechanism across multiple neurodegenerative diseases.
Neurons rely quasi-exclusively on mitochondria for ATP production, thus the selective clearance of damaged mitochondria by mitophagy is critical to maintain the bioenergetic integrity of the cell and prevent the accumulation of damaged mitochondria and toxic reactive oxygen species. The reliance on mitochondria also means mitophagy is a rare event, and most studies require overexpression and induction of mitophagy using mitochondrial uncoupling agents that artificially induce mitochondrial depolarization. A strength of this study is the cross-species analysis of basal mitophagy in multiple in vitro and in vivo models.
The work raises some interesting questions. There are multiple pathways coordinating mitochondrial integrity, and it will be important to identify the specific mitophagy pathway(s) that is/are dysfunctional in AD. The PINK1-dependent pathway is the most well-characterized mechanism for the induction and execution of mitophagy, however PINK1 knockout mice do not have defective mitophagy (McWilliams et al., 2018), and so it is intriguing that such widespread alterations in basal mitophagy are observed in AD models. The PINK1 pathway could be explored more extensively in these models by using alternative methods, such as mtQC mitophagy mice reporters (McWilliams et al., 2018), and by examining (for example) the levels of phosphorylated ubiquitin at serine 65, which is a substrate of PINK1 precluding the recruitment of Parkin to damaged mitochondria. It would also be important to study mitochondrial biogenesis in more detail, especially as PGC1a levels seem to be reduced in their AD models. Finally, it would be interesting to know whether the defects observed in hippocampus also occur in other brain regions in their disease models.
A number of studies have suggested an interplay of neuronal and glial cells to orchestrate mitophagy in a transcellular manner (Davis et al., 2014), yet this paper suggests a new role for mitophagy in enhancing a dual role for microglia: the phagocytic clearance of Aβ plaques and the release of pro-inflammatory cytokines. The role of the connected network of neurons and glial cells will need to be studied in more detail in AD and in other neurodegenerative conditions.
Together, these data add to a growing body of evidence highlighting the importance of mitophagy across multiple diseases. Future mechanistic studies will highlight the potential of this pathway for therapeutic targeting.
References:
McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017 Apr;45:83-91. Epub 2017 Apr 22 PubMed.
Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011 Aug;23(4):476-82. Epub 2011 Jun 24 PubMed.
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015 Oct 1;60(1):7-20. Epub 2015 Sep 10 PubMed.
Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013 Apr 10;78(1):65-80. PubMed.
Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017 Dec 14;552(7684):187-193. Epub 2017 Dec 6 PubMed.
Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain. 2017 Dec 1;140(12):3233-3251. PubMed.
Checler F, Goiran T, Alves da Costa C. Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: Implication for neurodegenerative diseases. Autophagy. 2017;13(11):2004-2005. Epub 2017 Sep 21 PubMed.
Martín-Maestro P, Gargini R, Perry G, Avila J, García-Escudero V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer's disease. Hum Mol Genet. 2016 Feb 15;25(4):792-806. Epub 2015 Dec 31 PubMed.
McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MM, Brooks SP, Ganley IG. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018 Feb 6;27(2):439-449.e5. Epub 2018 Jan 11 PubMed.
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014 Jul 1;111(26):9633-8. Epub 2014 Jun 16 PubMed.
Quietmind Foundation
Quietmind Foundation
We agree with Xiongwei Zhu and Wenzhang Wang on significant evidence supporting a critical role of mitochondrial dysfunction in Alzheimer's disease. We have pursued this premise to conduct IRB-approved placebo controlled clinical trials in human LOAD.
Our clinical experience and published findings on transcranial and intraocular infrared photobiomodulation suggests that increasing ATP activation through stimulating Cytochrome C Oxidase in the mitochondria results in improved cognitive and behavioral functioning in adults with dementia and Parkinson's disease. Recent AD animal model data showed this type of noninvasive stimulation improved praxis memory, motor behavior, reduced Aβ42 and p-tau, confers neuroprotection, and significantly increased VEGF production. Expanded human clinical trials are now underway at Baylor Research Institute (Temple, Texas) and Quietmind Foundation (Elkins Park, Pennsylvania) to evaluate the impact of self-administered, twice-daily, six-minute, transcranial and intraocular exposure to near infrared photobiomodulation.
Jason Huang also contributed to this comment.
References:
Berman MH, Halper JP, Nichols TW, Jarrett H, Lundy A, Huang JH. Photobiomodulation with Near Infrared Light Helmet in a Pilot, Placebo Controlled Clinical Trial in Dementia Patients Testing Memory and Cognition. J Neurol Neurosci. 2017;8(1) Epub 2017 Feb 28 PubMed.
Grillo SL, Duggett NA, Ennaceur A, Chazot PL. Non-invasive infra-red therapy (1072 nm) reduces β-amyloid protein levels in the brain of an Alzheimer's disease mouse model, TASTPM. J Photochem Photobiol B. 2013 Jun 5;123:13-22. Epub 2013 Mar 22 PubMed.
Duggett NA, Chazot PL. Low-Intensity Light Therapy (1068nm) Protects CAD Neuroblastoma Cells from β-Amyloid-Mediated Cell Death. Biol Med 2014
Lee SY, Seong IW, Kim JS, Cheon KA, Gu SH, Kim HH, Park KH. Enhancement of cutaneous immune response to bacterial infection after low-level light therapy with 1072 nm infrared light: a preliminary study. J Photochem Photobiol B. 2011 Dec 2;105(3):175-82. Epub 2011 Sep 6 PubMed.
Weill Cornell Medical College
Aging, the most important risk factor for AD, is linked to reductions in mitophagy. This is a remarkable and outstanding paper since it ties tau phosphorylation and amyloid pathology to mitochondrial dysfunction and defective mitophagy. These findings shed light on the underlying mechanisms of mitophagy and emphasize the importance of developing new therapeutic strategies targeting mitochondrial dysfunction in AD and other neurodegenerative diseases.
Cellular energy is mainly produced via mitochondrial oxidative phosphorylation (OXPHOS). Impaired mitochondrial function is a major contributor to human pathology and plays a critical role in the onset and progression of neurodegenerative diseases. Accumulation of misfolded proteins, downregulation of mitochondrial electron transport chain activity, axonopathy, and unbalanced mitochondrial dynamics are prevalent features of mitochondrial dysfunction.
Aberrant mitochondrial function has been linked to the pathogenesis of Alzheimer’s disease (AD), which is characterized by the formation of amyloid β (Aβ) plaques and hyperphosphorylated tau in the form of intraneuronal neurofibrillary tangles (Perez Ortiz and Swerdlow, 2019). Accumulation of dysfunctional mitochondria diminishes ATP production and disrupts the redox balance, which initiates oxidative damage, mitochondrial deficits, and subsequent amyloid pathology. Mitochondrial accumulation of Aβ and APP (mediated by the TOM-TIM protein complexes) was detected in the frontal cortices and hippocampi of AD patients and transgenic AD mice (Anandatheerthavarada et al., 2003; Lustbader et al., 2004; Devi et al., 2006). Overexpression of Aβ resulted in the perturbation of mitochondrial dynamics with concomitant abnormal mitochondrial morphology and distribution in vitro and in Tg2576 mice (Wang et al., 2008; Calkins et al., 2011). Changes in the concentration of the mitochondrial fission and fusion-related proteins, morphological structure, and localization have been reported in fibroblasts and subjects with sporadic AD (Wang et al., 2008; Manczak et al., 2011; Martin-Maestro et al., 2017). Furthermore, an early and progressive accumulation of Aβ (mainly Aβ1-42) within mitochondria leads to synaptic mitochondrial dysfunction in transgenic mice overexpressing APP/Aβ (Du et al., 2010).
Selective degradation of mitochondria by autophagy (mitophagy) is part of the quality-control system of cells and is essential to maintain mitochondrial homeostasis. Disruption of mitophagy results in mitochondrial membrane depolarization, ATP depletion, and oxidative damage. PTEN-induced putative kinase 1 (PINK1) accumulates on dysfunctional mitochondria and its kinase activity is required for the translocation to mitochondria of parkin (a cytosolic E3 ubiquitin) to mediate the autophagic removal of damaged mitochondria. Mutations in PINK1 and parkin prevent PINK1 recruitment of parkin into mitochondria and are responsible for autosomal-recessive and some sporadic forms of Parkinson's disease (PD). Clinical and experimental evidence has demonstrated that mitophagy is compromised in AD. Parkin-mediated mitophagy was significantly activated in cultured cortical neurons derived from human APP transgenic mice and postmortem hippocampal tissue specimens from AD patients (Ye et al., 2015).
Which are the effects of tau and Aβ on mitophagy quality control? Impaired mitophagy was observed in N2a cells transfected with wild-type and P301L mutant human tau and transgenic C. elegans (Cummins et al., 2019). Expression of tau also disrupted the translocation of Parkin to mitochondria through a mitochondrial membrane potential-independent mechanism. Aβ accumulation in synaptosomal mitochondria of 5xFAD mice decreased axonal mitochondrial length, altered mitochondrial dynamics, and increased Parkin-mediated mitophagy (Wang et al., 2016). Overexpression into the hippocampi of mice of either PINK1 or parkin, the primary enzymes implicated in mediating mitophagy, can exert beneficial effects on the neuropathological hallmarks of AD, Aβ plaques, and neurofibrillary tangles (Khandelwal et al., 2011; Du et al., 2017). Perturbed mitochondrial proteostasis and activated mitophagy have been reported in transgenic C. elegans, 3xTgAD mice, and brain tissue from AD patients. Treatment with nicotinamide riboside, which increases NAD+ levels and activates sirtuins, significantly reduced Aβ aggregates and associated proteotoxicity (Sorrentino et al., 2017). More recently, in a well-conducted study, Bohr’s group showed alterations in mitochondrial function and changes in the immunoreactivity of mitophagy-associated proteins in both brain biopsies and iPSC-derived neurons from AD patients (Fang et al., 2019). The levels of 5' AMP-activated protein kinase, which leads to excessive mitochondrial fission and ATP depletion, were increased in the hippocampi of patients with AD. They showed that pharmacologic or genetic activation of mitophagy significantly mitigated cognitive deficits and Aβ-induced toxicity in APP/PS1 AD mice. Mitophagy induction also reduced tau hyperphosphorylation and improved memory deficits and cognitive decline in nematodes and mice, respectively. Pharmacological restoration of mitophagy promoted microglial phagocytosis and mitigated neuroinflammation in a double-transgenic mouse model of AD.
References:
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003 Apr 14;161(1):41-54. PubMed.
Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011 Dec 1;20(23):4515-29. PubMed.
Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Götz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019 Feb 1;38(3) Epub 2018 Dec 11 PubMed.
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006 Aug 30;26(35):9057-68. PubMed.
Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain. 2017 Dec 1;140(12):3233-3251. PubMed.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2010 Oct 26;107(43):18670-5. PubMed.
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019 Mar;22(3):401-412. Epub 2019 Feb 11 PubMed.
Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011 Jun 1;20(11):2091-102. PubMed.
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004 Apr 16;304(5669):448-52. PubMed.
Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011 Jul 1;20(13):2495-509. PubMed.
Martín-Maestro P, Gargini R, García E, Perry G, Avila J, García-Escudero V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer's Disease. Oxid Med Cell Longev. 2017;2017:9302761. Epub 2017 Oct 19 PubMed. Correction.
Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019 Jan 24; PubMed.
Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017 Dec 14;552(7684):187-193. Epub 2017 Dec 6 PubMed.
Wang L, Guo L, Lu L, Sun H, Shao M, Beck SJ, Li L, Ramachandran J, Du Y, Du H. Synaptosomal Mitochondrial Dysfunction in 5xFAD Mouse Model of Alzheimer's Disease. PLoS One. 2016;11(3):e0150441. Epub 2016 Mar 4 PubMed.
Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008 Aug;173(2):470-82. PubMed.
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008 Dec 9;105(49):19318-23. PubMed.
Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet. 2015 May 15;24(10):2938-51. Epub 2015 Feb 12 PubMed.
The University of Queensland
We share the view of Drs. Tapias and Beal that misfolded proteins such as Aβ and tau impair all aspects of mitochondrial functions. In fact, we have a long-standing interest in how Aβ and tau impair neuronal functions, dating back to 2005 when we combined a proteomic with a functional analysis showing impairments to the OXPHOS system (David et al., 2005).
What is relevant in the context of how mitochondrial function is impaired under pathological conditions is a study which we recently performed that adds another layer to mitochondrial dysfunctions and how the cell responds to it. We developed a protocol in primary hippocampal cultures that combines the photosensitizer mito-KillerRed with fluorescent biosensors and photoactivatable GFP. We found in both the soma and dendrites that by quarantining mitochondria, neurons restrict the local increase in mitochondria-derived reactive oxygen species and the decrease in ATP production to the damaged compartment.
Interestingly, although the cytosol of both the soma and dendrites became oxidized after mito-KillerRed activation, dendrites were more sensitive to the oxidative insult. Importantly, the impaired mitochondria exhibited decreased motility and fusion, thereby avoiding the spread of oxidation throughout the neuron. These results establish how neurons manage oxidative damage and increase our understanding about the somatodendritic regulation of mitochondrial function after a local oxidative insult (Grimm et al., 2018).
References:
David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Müller WE, Eckert A, Götz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005 Jun 24;280(25):23802-14. PubMed.
Grimm A, Cummins N, Götz J. Local Oxidative Damage in the Soma and Dendrites Quarantines Neuronal Mitochondria at the Site of Insult. iScience. 2018 Aug 31;6:114-127. Epub 2018 Jul 23 PubMed.
Make a Comment
To make a comment you must login or register.