Clotting Protein from Blood Incites Microglia, and Synapses Die
Quick Links
A damaged and leaky blood-brain barrier contributes to dementia, but just how has not been clear. Previous work implicated the clotting protein fibrinogen, which, when it penetrates the brain and coagulates, activates microglia and leads to inflammation and neurodegeneration. Now, Katerina Akassoglou’s group at the Gladstone Institutes, University of California, San Francisco, shows that in a mouse model of amyloid deposition, fibrinogen deposits incite microglia to destroy synapses, leading to cognitive decline. Synapse loss did not require amyloid. Preventing fibrinogen binding to the CD11b receptor on microglia improved cognition in the mice. The work appeared February 5 in Neuron.
- Once inside the brain, fibrinogen from blood induces synapse loss.
- In AD transgenic mice, it causes cognitive decline.
- Blocking interaction between fibrinogen and microglial receptor improves cognition.
“This interesting work shows that fibrinogen deposition is a mechanism linking microglia activation/neuroinflammation and neuronal dysfunction in mouse models of Alzheimer’s disease. Importantly, the results suggest that targeting fibrinogen might be therapeutically beneficial in some cases of AD,” said Sidney Strickland of Rockefeller University in New York.
In older people, damage to blood vessels in the brain and amyloid burden independently and additively predict cognitive decline (Jan 2019 news). Scientists know that Aβ oligomers cause synaptic pruning by microglia (Apr 2016 news). The new findings suggest fibrinogen and Aβ activate parallel immune responses that lead to synapse loss. “These findings are in harmony with a new recognition of Alzheimer’s disease as a multifactorial disease with multiple contributors to its pathophysiology, including vascular dysfunction,” wrote Axel Montagne, Keck School of Medicine, University of Southern California, Los Angeles (see comment below).
As the Akassoglou lab previously discovered, when blood leaks into the brain, cleavage of fibrinogen by thrombin uncovers a cryptic binding site for the CD11b receptor on microglia. Through that receptor, fibrinogen stimulates inflammation and the release of toxic free radicals. A monoclonal antibody that blocks the fibrinogen-CD11b interaction prevented neurodegeneration in mouse models of AD or multiple sclerosis, they showed (Oct 2018 news).
In the new study, authors Mario Merlini, Victoria Rafalski, and Pamela Rios Coronado took a closer look at brain fibrinogen in 5xFAD mice, a model of rapid amyloid accumulation, and in human AD brain. Using a polyclonal antibody to fibrinogen, they detected immunoreactivity in some cortical amyloid plaques, but not all. They also found amyloid-free fibrinogen deposits peppered throughout the mouse cortex. In human brain tissue, parenchymal deposits of fibrinogen were more frequent in samples from people with AD than in age-matched, nondemented controls, and appeared even in areas free of Aβ plaques.
Repeated two-photon imaging in living mice revealed progressive dendritic spine loss around Aβ plaques, as expected. The loss occurred whether fibrinogen was present or not. Surprisingly, spines were lost around fibrinogen-only deposits as well, even when they were far from Aβ plaques.
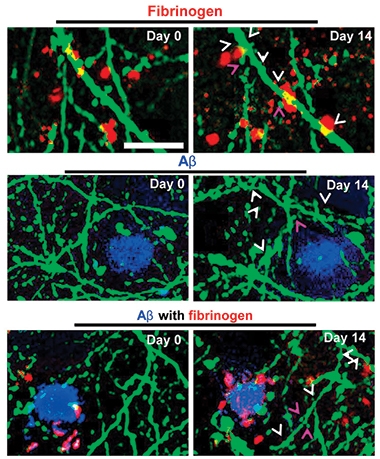
Lost Connections. In 5xFAD mice, neurites (green) near fibrinogen deposits (red), Aβ plaques (blue) or mixed deposits all lose spines over time. White arrows indicate spots where spines disappeared after 14 days; magenta arrowheads mark stable spines. [Courtesy of Neuron, Merlini et al., 2019.]
To determine whether fibrinogen alone could damage dendrites, the scientists injected purified human or mouse fibrinogen into the brains of healthy mice. Three days later, they detected localized dendrite and spine loss around the injection sites. Injecting whole plasma, a source of abundant fibrinogen, also caused spine loss, but plasma from fibrinogen-knockout mice did not. Spine loss induced by fibrinogen required CD11b, as injecting fibrinogen had no effect in CD11b-knockout mice. In the brain, microglia exposed to fibrinogen assumed an activated morphology and produced reactive oxygen species (ROS) that damaged neurons. An ROS inhibitor reduced spine loss and partially preserved dendrites in the mice.
“Our results suggest that cerebral extravasation of blood proteins causes neurite loss and that fibrinogen is a major mediator of this effect,” said Akassoglou. “We can now start connecting the dots between blood leaks in the brain, activation of the immune system, and the elimination of neuronal connections that are important for memory and cognition,” she said.
What about cognition in the mice that were losing spines? To find out, the investigators crossed the 5xFAD mice with mice expressing a mutated fibrinogen that did not bind CD11b. Whereas 10-month-old 5xFAD mice had severe trouble remembering and avoiding an unpleasant stimulus, the 5xFAD/mutant fibrinogen mice remembered almost as well as wild-type mice. They also showed little penchant for hyperactivity, another hallmark of 5xFAD mice.
The 5xFAD/fibrinogen mutant mice had up to 50 percent higher neuronal and synaptic markers in the dentate gyrus, and fewer hippocampal Aβ plaques. By zeroing in on CD11b binding as the critical interaction, the results expand previous work from the Strickland lab, indicating fibrinogen knockout in AD mice improved cognition by reducing dystrophic neurites, lowering amyloid plaque load, and preventing neurodegeneration (Cortes-Canteli et al., 2015; Cortes-Canteli et al., 2010).
“It would be of interest to elucidate at which stage of the disease process this mechanism is active. Is it a terminal event or an early driver of pathology and dysfunction?” asked Costantino Iadecola in a message to Alzforum. “Inasmuch as the blood-brain barrier permeability increase is an early event in AD, the data would suggest that targeting this mechanism may be beneficial, as suggested by the cognitive benefit observed in the mouse model,” he added (see comment below).—Pat McCaffrey
References
News Citations
- Absent Aβ, Blood-Brain Barrier Breakdown Predicts Cognitive Impairment
- Paper Alert: Microglia Mediate Synaptic Loss in Early Alzheimer’s Disease
- Getting Between Fibrin and Complement Receptor Cools Inflammation
Research Models Citations
Paper Citations
- Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging. 2015 Feb;36(2):608-17. Epub 2014 Oct 31 PubMed.
- Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM, Strickland S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010 Jun 10;66(5):695-709. PubMed.
Further Reading
Papers
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, Markus HS, Salman RA, Allan SM, Quinn TJ, Kalaria RN, Werring DJ, Carare RO, Touyz RM, Williams SC, Moskowitz MA, Katusic ZS, Lutz SE, Lazarov O, Minshall RD, Rehman J, Davis TP, Wellington CL, González HM, Yuan C, Lockhart SN, Hughes TM, Chen CL, Sachdev P, O'Brien JT, Skoog I, Pantoni L, Gustafson DR, Biessels GJ, Wallin A, Smith EE, Mok V, Wong A, Passmore P, Barkof F, Muller M, Breteler MM, Román GC, Hamel E, Seshadri S, Gottesman RF, van Buchem MA, Arvanitakis Z, Schneider JA, Drewes LR, Hachinski V, Finch CE, Toga AW, Wardlaw JM, Zlokovic BV. Vascular dysfunction-The disregarded partner of Alzheimer's disease. Alzheimers Dement. 2019 Jan;15(1):158-167. PubMed. Correction.
- Petersen MA, Ryu JK, Akassoglou K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci. 2018 May;19(5):283-301. Epub 2018 Apr 5 PubMed.
Primary Papers
- Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, Seeley WW, Mucke L, Nelson RB, Akassoglou K. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer's Disease Model. Neuron. 2019 Mar 20;101(6):1099-1108.e6. Epub 2019 Feb 5 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Weill College Medicine, New York
This elegant study closes the loop between BBB disruption, extravasation of fibrinogen, and cognitive dysfunction in a mouse model of amyloid accumulation. It was known that fibrinogen enters the brain through a breach of the BBB and that the interaction of fibrinogen with the immune receptor CD11b contributed to the cognitive dysfunction seen in these models, but the ultimate effector of the dysfunction remained unclear. The evidence unveiled a role for synaptic pruning in the fibrinogen-induced cognitive dysfunction. These data are relevant because they (a) identify a specific mechanism for the pathogenic effect of BBB opening in models of amyloid accumulation, and, perhaps, Alzheimer’s disease, (b) point to synaptic pruning as a key effector of the dysfunction, and (c) identify the CD11b-fibrinogen interaction as a potential therapeutic target.
Since CD11b is present in microglia, a cell type involved in spine remodeling and pruning, the data strongly suggest that the interaction of fibrinogen with microglia is critical. However, this receptor is also present in resident brain macrophages located in the meninges, perivascular space, and choroid plexus. We have linked resident brain macrophages to the neurovascular dysfunction induced by amyloid-β peptides (Park et al., 2017) and due to their location in the perivascular space these would be one of the first cells engaged by fibrinogen escaping the vessels and entering the brain. The observation that the free-radical-producing enzyme NADPH oxidase is enriched particularly in brain macrophages is also consistent with the protective effect of the NADPH oxidase inhibitor apocynin. Further studies in which CD11b is selectively silenced either in microglia or macrophages would be needed to address the CD11b-expressing cell type(s) involved in spine pruning.
As for the translational implications of the findings, if would be of interest to elucidate at which stage of the disease process this mechanism is active. Is it a terminal event or an early driver of pathology and dysfunction? Inasmuch as the BBB permeability increase is an early event in AD (Montagne et al., 2015; Nation et al., 2019), the data would suggest that targeting this mechanism may be beneficial, as suggested by the cognitive benefit observed in the mouse model. In this regard antibody treatment targeting the CD11b-fibrinogen interaction would be of interest (Ryu et al., 2018).
References:
Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, Younkin L, Younkin S, Zhou P, Carlson G, Anrather J, Iadecola C. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ Res. 2017 Jul 21;121(3):258-269. Epub 2017 May 17 PubMed.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015 Jan 21;85(2):296-302. PubMed.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TL, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019 Feb;25(2):270-276. Epub 2019 Jan 14 PubMed.
Ryu JK, Rafalski VA, Meyer-Franke A, Adams RA, Poda SB, Rios Coronado PE, Pedersen LØ, Menon V, Baeten KM, Sikorski SL, Bedard C, Hanspers K, Bardehle S, Mendiola AS, Davalos D, Machado MR, Chan JP, Plastira I, Petersen MA, Pfaff SJ, Ang KK, Hallenbeck KK, Syme C, Hakozaki H, Ellisman MH, Swanson RA, Zamvil SS, Arkin MR, Zorn SH, Pico AR, Mucke L, Freedman SB, Stavenhagen JB, Nelson RB, Akassoglou K. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol. 2018 Nov;19(11):1212-1223. Epub 2018 Oct 15 PubMed.
University of Edinburgh
Akassoglou’s lab has published a very elegant study demonstrating that blood-derived fibrinogen that leaks out into the brain is synaptotoxic and leads to cognitive impairment independently of amyloid pathology. The authors also found that fibrin(ogen) at sites of blood-brain barrier (BBB) damage can physically interact with CD11b/CD18 integrin microglial receptors (a.k.a. complement receptor 3) which, in turn, causes microglial activation and oxidative stress leading to dendrite and spine losses. The authors used well-designed pharmacological and genetical approaches to strengthen their conclusion.
These findings are in accordance with our recent study in pericyte-deficient mice showing that early fibrin(ogen) deposits in white matter trigger pericytes’ and oligodendrocytes’ death by autophagy, ultimately leading to axon and myelin damage followed by neuronal loss and cognitive dysfunction (Montagne et al., 2018). Merlini’s study and previous studies from the same group demonstrate that fibrin promotes neuroinflammation and microglia activation. In contrast, we failed to detect changes in the number of white-matter reactive astrocytes and activated microglia in young, middle-aged, and old pericyte-deficient mice or changes in cytokine and chemokine expression levels (Montagne et al., 2018). Regardless, it is now conspicuous that fibrin(ogen) is toxic for both white and grey matters through different mechanisms involving different cell types.
Importantly, these findings are in harmony with this new era recognizing Alzheimer's disease (AD) as a multifactorial disease with multiple contributors to its pathophysiology, including vascular dysfunction (Sweeney et al., 2019). Recently, our group showed that people with a damaged BBB were more likely to have early signs of cognitive impairment, regardless of any amyloid-β (Aβ) or tau pathology, or even other signs of vascular disease (Nation et al., 2019). Akassoglou’s group also suggest in their Neuron article that vascular pathology is independent of, but additive to, Aβ load in mediating cognitive decline.
How BBB dysfunction promotes AD remains to be elucidated, but evidence for the pro-inflammatory or pro-cytotoxic effects of fibrin(ogen) entering the brain has been proposed in the present study and by others. It has considerable therapeutic implications in AD patients and other neurodegenerative diseases (e.g., multiple sclerosis, small-vessel disease, or CADASIL). One treatment approach would be to use 5B8 monoclonal antibody developed by the same group, which would block fibrin(ogen)-complement receptor 3 interaction, alleviating the innate immune response while leaving fibrin’s platelet-interacting site unperturbed for proper coagulation cascade. Another possibility would be to act upstream by sealing the BBB, thus preventing pro-cytotoxic fibrin(ogen) molecules from entering the brain. One candidate to be considered is the genetically engineered protein 3K3A-activated protein C (APC), which was shown to be safe, well-tolerated, and capable of reducing intracerebral bleeding after stroke in humans (Lyden et al., 2019). The vasculoprotective effect of 3K3A-APC could also be explored in AD patients, as a recent study in 3K3A-APC-treated AD mice revealed improved BBB integrity, Aβ clearance, and cognition (Lazic et al., 2019).
Given the recent advances in the field of microbiome research, it would be interesting to study other potential CD11b ligands including, but not limited to, oral bacterial pathogens which might also intervene in this pro-inflammatory cascade and influence Aβ clearance (Olsen and Singhrao, 2015; Dominy et al., 2019).
References:
Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, Barnes SR, Daianu M, Ramanathan A, Go A, Lawson EJ, Wang Y, Mack WJ, Thompson PM, Schneider JA, Varkey J, Langen R, Mullins E, Jacobs RE, Zlokovic BV. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med. 2018 Mar;24(3):326-337. Epub 2018 Feb 5 PubMed. RETRACTED
Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, Markus HS, Salman RA, Allan SM, Quinn TJ, Kalaria RN, Werring DJ, Carare RO, Touyz RM, Williams SC, Moskowitz MA, Katusic ZS, Lutz SE, Lazarov O, Minshall RD, Rehman J, Davis TP, Wellington CL, González HM, Yuan C, Lockhart SN, Hughes TM, Chen CL, Sachdev P, O'Brien JT, Skoog I, Pantoni L, Gustafson DR, Biessels GJ, Wallin A, Smith EE, Mok V, Wong A, Passmore P, Barkof F, Muller M, Breteler MM, Román GC, Hamel E, Seshadri S, Gottesman RF, van Buchem MA, Arvanitakis Z, Schneider JA, Drewes LR, Hachinski V, Finch CE, Toga AW, Wardlaw JM, Zlokovic BV. Vascular dysfunction-The disregarded partner of Alzheimer's disease. Alzheimers Dement. 2019 Jan;15(1):158-167. PubMed. Correction.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TL, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019 Feb;25(2):270-276. Epub 2019 Jan 14 PubMed.
Lyden P, Pryor KE, Coffey CS, Cudkowicz M, Conwit R, Jadhav A, Sawyer RN Jr, Claassen J, Adeoye O, Song S, Hannon P, Rost NS, Hinduja A, Torbey M, Lee JM, Benesch C, Rippee M, Rymer M, Froehler MT, Clarke Haley E, Johnson M, Yankey J, Magee K, Qidwai J, Levy H, Mark Haacke E, Fawaz M, Davis TP, Toga AW, Griffin JH, Zlokovic BV, NeuroNEXT Clinical Trials Network NN104 Investigators. Final Results of the RHAPSODY Trial: A Multi-Center, Phase 2 Trial Using a Continual Reassessment Method to Determine the Safety and Tolerability of 3K3A-APC, A Recombinant Variant of Human Activated Protein C, in Combination with Tissue Plasminogen Activ. Ann Neurol. 2019 Jan;85(1):125-136. Epub 2019 Jan 7 PubMed.
Lazic D, Sagare AP, Nikolakopoulou AM, Griffin JH, Vassar R, Zlokovic BV. 3K3A-activated protein C blocks amyloidogenic BACE1 pathway and improves functional outcome in mice. J Exp Med. 2019 Feb 4;216(2):279-293. Epub 2019 Jan 15 PubMed.
Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer's disease?. J Oral Microbiol. 2015;7:29143. Epub 2015 Sep 17 PubMed.
Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RL, Curtis MA, Dragunow M, Potempa J. Porphyromonas gingivalis in Alzheimer's disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019 Jan;5(1):eaau3333. Epub 2019 Jan 23 PubMed.
University of Gothenburg
We and others regularly observe a differential profile of blood fibrinogen being associated with brain amyloid pathology and cognitive decline in Alzheimer’s disease patients, without fully understanding its mechanistic importance. Previous reports have shown that fibrinogen crosses the blood-brain barrier to co-deposit with amyloid plaques and activated microglia. Now, we see an Aβ-independent cause of cognitive decline that is seemingly provoked by the influx of fibrinogen from the periphery. Therapies directed toward specific fibrin interactions could be of great benefit to the large proportion of those with a vascular contribution to cognitive decline.
University College London
The finding that blood-derived fibrinogen has direct effects on brain microglia and dendritic spines provides an intriguing mechanism by which cerebrovascular disease may influence cognition. This work tallies well with recent findings that blood brain barrier breakdown is associated with cognitive impairment independent of Aβ and tau pathology (Jan 2019 news), and postmortem studies demonstrating that extravascular fibrinogen relates to cerebrovascular disease and not AD pathology (McAleese et al., 2018).
While these findings need to be explored further in living patients, e.g., with CSF measures of fibrinogen, they add to a growing body of evidence that blood-brain compromise can lead to cognitive impairment which may or may not relate to, or be synergistic with, Alzheimer’s disease, but where it does, it is seemingly independent of classical Aβ and tau pathology.
References:
McAleese KE, Graham S, Dey M, Walker L, Erskine D, Johnson M, Johnston E, Thomas AJ, McKeith IG, DeCarli C, Attems J. Extravascular fibrinogen in the white matter of Alzheimer's disease and normal aged brains: implications for fibrinogen as a biomarker for Alzheimer's disease. Brain Pathol. 2019 May;29(3):414-424. Epub 2019 Jan 29 PubMed.
UC Irvine, MIND Institute and Center for Neuroscience of Memory
We were glad to see Melini et al. show in whole animals an integrated anatomical, physiologic, and cognitive effect of fibrinogen leaks from the plasma into the parenchyma. Now many directions of research confirm what we suspected when we showed how thrombin affects neurons directly in our 1996 J. Neurochem. paper.
References:
Brewer GJ. Thrombin causes cell spreading and redistribution of beta-amyloid immunoreactivity in cultured hippocampal neurons. J Neurochem. 1996 Jul;67(1):119-30. PubMed.
View all comments by Gregory BrewerGachon University
We support the report from Melini et al.
Fibrinogen will attract other molecules, especially tissue plasminogen activator (tPA) and plasminogen. In addition, the activation of tPA in brain was shown to be very toxic to neurons (Kaur et al., 2004; Liu et al., 2004); this could be the reason for the degradation of synapses.
Lastly, we recently published a related report on the potential direct binding between β-amyloid and fibrinogen as following, "Epitope mapping immunoassay analysis of the interaction between β-amyloid and fibrinogen" (Van Giau et al., 2019.)
References:
Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator?. J Cereb Blood Flow Metab. 2004 Sep;24(9):945-63. PubMed.
Liu D, Cheng T, Guo H, Fernández JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004 Dec;10(12):1379-83. Epub 2004 Oct 31 PubMed.
Van Giau V, An SS. Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen. Int J Mol Sci. 2019 Jan 24;20(3) PubMed.
Make a Comment
To make a comment you must login or register.