Electron Microscope Yields Finer Structure of α-Synuclein, Aβ Fibrils
Quick Links
Protein aggregates associated with neurodegenerative disease have stubbornly resisted researchers’ efforts to get a good look at them. They refuse to crystallize well or yield to standard spectroscopic techniques. Now, advances in electron microscopy methods are forcing these molecules to give up their secrets. In the September 9 Nature, researchers led by David Eisenberg at the University of California, Los Angeles, and Tamir Gonen at the Howard Hughes Medical Institute’s Janelia Research Campus in Ashburn, Virginia, offer the closest look yet at the core of α-synuclein aggregates. The researchers made microscopic crystals from the peptides and used a relatively new technique called micro-electron diffraction to map them down to atomic resolution. Commenters called the work a tour de force.
“[These] structures are the first to be determined by micro-electron diffraction from a molecule of previously unknown structure,” noted Yifan Cheng at the University of California, San Francisco, in an accompanying Nature commentary. Others were similarly impressed. “To obtain a structure of this quality from a peptide material with such tiny crystals is a remarkable feat, and will probably serve as the model for many other studies,” Gregory Petsko at Weill Cornell Medical College in New York told Alzforum. Tim Bartels at Brigham and Women’s Hospital, Boston, agreed, “The resolution here is unprecedented.”

β-Sheet Sandwich.
The core portion of α-synuclein forms β strands that pair up (top), then stack to make β sheets (bottom). [Courtesy of Rodriguez et al., Nature.]
Meanwhile, in the September 8 Proceedings of the National Academy of Sciences, researchers led by Nikolaus Grigorieff at Janelia and Marcus Fändrich at Ulm University, Germany, describe a detailed structure for Aβ42 fibrils using electron cryomicroscopy. While cryoEM’s resolution is a tad lower than micro-electron diffraction's, this method allowed the researchers to examine the aggregated peptide in a more natural, hydrated state. Their model jibes with previous structural findings and explains some of the observed behavior of amyloid fibrils, commenters said.
It remains unknown whether the structures described in these papers occur in human disease. Researchers know that proteins aggregate into different strains, so the solved structures may represent but one possible configuration among many. Nonetheless, scientists expect these and future models to advance drug discovery. “The data will give medicinal chemists targets for designing therapeutic compounds,” noted David Teplow at UCLA. He was not involved in the work.
For many proteins, researchers obtain high-resolution structural data using nuclear magnetic resonance (NMR) spectroscopy or X-ray crystallography. However, aggregated neurodegenerative proteins do not easily form the large crystals, or homogenous aggregates, these methods require. α-Synuclein, which accumulates in Parkinson’s disease, multiple-system atrophy (MSA), and other disorders, has been particularly recalcitrant. Previous spectroscopy studies hinted that the peptide contains β strands and that its fibril likely consists of β sheets, but the finer points were lacking (see Heise et al., 2005; Chen et al., 2007; Cho et al., 2011).
To get a closer look, Eisenberg and colleagues turned to micro-electron diffraction, a technique that combines features of X-ray crystallography and cryoEM. As in cryoEM, samples are frozen in a hydrated state, preserving their structure, then subjected to a beam of electrons in a transmission electron microscope. Unlike cyroEM, micro-electron diffraction does not directly produce an image of the sample, but instead analyzes a diffraction pattern, as does X-ray crystallography. This allows researchers to probe minute crystals, 10 billion times smaller than some of those used in X-ray crystallography, Eisenberg noted. By rotating these tiny crystals in the electron beam, researchers collect a series of diffraction patterns that reveal the three-dimensional structure of the protein. Previously, this method had been applied only to confirm known protein configurations.
Because full-length α-synuclein resists crystallization, joint first authors Jose Rodriguez, Magdalena Ivanova, Michael Sawaya, and Duilio Cascio at UCLA and Francis Reyes at Janelia crystallized a small segment comprising residues 68-78. This portion forms the core of a longer region known as NAC, or non-amyloid-β component. Other researchers have reported that this short “NACore,” as the authors call it, forms toxic amyloid fibrils on its own, while deleting the segment from α-synuclein prevents aggregation (see El-Agnaf et al., 1998; Periquet et al., 2007). The data imply that this NACore region controls aggregation of the full-length protein, the authors note. They aggregated a synthetic NACore fragment in vitro, obtaining crystals 50-300 nm across. Micro-electron diffraction then allowed them to visualize these crystals down to a resolution of 1.4 Å.
Diffraction data from the crystals supported a model in which two NACore strands dimerize tightly together, then stack up in layers to form two facing β sheets, similar to the configuration of other amyloid aggregates (see image above). Layers were separated by 4.8 Å and were staggered from each other, matching predictions from previous models. Surprisingly, however, the researchers found two water molecules nestled between the sheets in each layer. Most other β-sheet structures are dry. “This might imply that these α-synuclein fibers are not as ultra-stable as some other fibers, like Aβ,” Eisenberg suggested.

Parkinson’s Mutation May Enhance Binding.
Computer modeling suggests that the A53T familial mutation leads to an additional β-sheet interface, perhaps promoting aggregation. [Courtesy of Rodriguez et al., Nature.]
The researchers also crystallized and analyzed a separate segment of α-synuclein comprising residues 47-56 and containing the A53T mutation associated with early onset Parkinson’s disease. The mutated segment by itself formed stacked β sheets. Computer modeling predicted that in the full-length protein, the mutated 47-56 segment would fold over the NACore β sheets, making a thicker sandwich (see image at left). This additional binding interface might explain why α-synuclein containing the mutation aggregates more readily than wild-type versions, the authors suggested. Other familial Parkinson’s mutations, such as A30P and E46K, lie outside the studied regions. “[That] suggests that further structural surprises may be in store,” wrote Michel Goedert at the MRC Laboratory of Molecular Biology, Cambridge, U.K., in a Nature commentary.
How closely does the structure of the aggregated NACore mimic that of fibrils composed of full-length protein, as seen by others? Diffraction patterns from the two aggregates look roughly similar, hinting that the structures may be comparable. However, commenters noted that additional β strands present in the full-length protein might affect folding. In ongoing work, Eisenberg and colleagues are attempting to crystallize longer segments of α-synuclein to get a fuller picture of the fibrils.
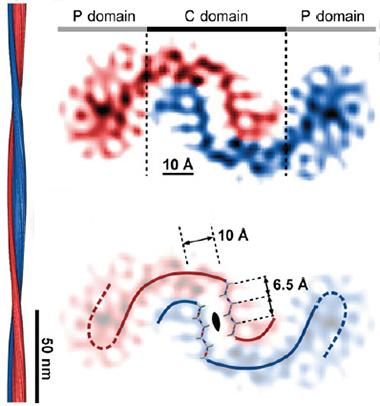
Tilde Turns?
CryoEM of Aβ42 suggests that the C-terminal domains of pairs of peptides bind in a curvy wave, while the N-terminal regions (P) splay out sideways. Computer modeling shows the presence of a steric zipper in the center of the binding interface (bottom). The fibril twists along its axis (left). [Courtesy of Schmidt et al., PNAS.]
How does this structure relate to reports of different structures or strains of α-synuclein (see Bousset et al., 2013; Peelaerts et al., 2015)? Ronald Melki at the French National Center for Scientific Research in the Paris suburb of Gif–sur–Yvette, noted that Eisenberg’s NACore structure matches the “ribbon” configuration of α-synuclein he recently described, which appears to associate more with MSA than PD (see Jun 2015 news; Sep 2015 news).
New Look at Aβ Fibrils
Grigorieff, Fändrich, and colleagues took a different tack to elucidate the structure of Aβ fibrils. They used standard cryoEM to bring fibrils into sharper focus than previous studies had allowed, achieving a resolution of about 7 Å. CryoEM does not require crystals; instead, researchers average images of several fibrils grown from the full-length peptide to build up a picture of the structure. Previously, the researchers used this technique to visualize Aβ40 fibrils. Those studies revealed structures composed of two protofilaments, each containing a paired β sheet at its core (see Sachse et al., 2008; Schmidt et al., 2009).
In the current study, joint first authors Matthias Schmidt at Ulm and Alexis Rohou at Janelia analyzed one type of synthetic Aβ42 aggregate made in vitro. A density map revealed two curved strands with their middle portions curling around each other (see image above). Computer modeling predicted that this structure was formed by the hydrophobic C-terminal domains of two Aβ42 peptides binding together in β strands, the side chains of their amino acids interdigitating in a “steric zipper.” Eisenberg had previously predicted this steric zipper based on X-ray crystallography of a small portion of Aβ, but not in the full fibril (see Sawaya et al., 2007).
Meanwhile, the charged N-terminal portions of each peptide splayed out to the sides. They were fuzzier, less resolved than the center, suggesting they move and assume multiple shapes. This jibes with previous data (see Dec 2010 conference news). The paired β strands of the peptides stack up to form sheets, composing the long axis of the fibril. The authors measured 10 Å between sheets, also in agreement with previous findings.
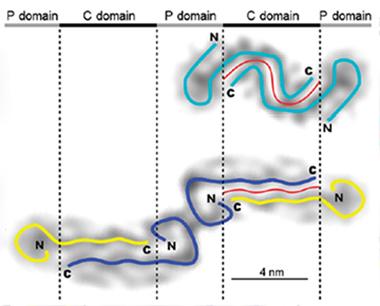
Distinct Shapes.
Aβ42 appears to form a single curvy protofilament (top), while Aβ40 fibrils consist of two straight protofilaments (bottom). [Courtesy of Schmidt et al., PNAS.]
Other features conflicted with previous models. A recent NMR study, led by Yoshitaka Ishii at the University of Illinois in Chicago, reported that Aβ42 monomers fold up on themselves in an S-shape to form the core of amyloid fibrils (see May 2015 news). By contrast, Grigorieff and Fändrich’s model places an Aβ dimer at the heart of the fibril, with each peptide assuming a gently curving tilde structure in place of sharp S-bends. It is possible these models simply represent two different strains of fibril produced under distinct aggregation conditions, commenters noted. Likewise, a third model of the Aβ42 fibril, obtained by cryoEM, depicts two intertwined protofilaments around a hollow core (see Mar 2009 news). These latter fibrils were incubated at a highly acidic pH, unlike the physiological conditions used by Grigorieff and Fändrich.
Which, if any, of these structures predominate in Alzheimer’s brains remains to be determined. However, the new tilde-shaped model “accounts for a lot of prior observations [about Aβ], and follows basic principles of protein folding,” Teplow noted. For example, the greater propensity of Aβ42 than Aβ40 to aggregate could be attributed to the longer interface between β strands in the former (see image above). Also, the distinct shapes of the Aβ40 and Aβ42 fibrils could explain why the two peptides do not readily form mixed fibrils, the authors suggest.
The new model predicts that two charged residues at the edges of the dimer interface, Glu693 and Asp694, help keep the ends of the strands apart and prevent stronger binding. Mutations of these residues that remove the charge, such as the E693G Arctic or the D694N Iowa mutation, lead to early onset AD, perhaps due to increased aggregation. This suggests that the fibril shares some structural features with toxic peptide aggregates, Grigorieff noted. “The link between fibril structure and familial Alzheimer’s mutations hints that fibrils may play an important role in the disease,” he said.—Madolyn Bowman Rogers
References
News Citations
- Shape of α-Synuclein Aggregates Influences Pathology
- α-Synuclein from Multiple System Atrophy Acts Like Prion in Mice
- San Diego: Flexible N-Termini Key to Aβ42 Oligomer Toxicity?
- Danger, S-Bends! New Structure for Aβ42 Fibrils Comes into View
- CryoEM Exposes Possible Achilles’ Heel in Aβ1-42 Fibrils
Mutations Citations
Paper Citations
- Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005 Nov 1;102(44):15871-6. Epub 2005 Oct 24 PubMed.
- Chen M, Margittai M, Chen J, Langen R. Investigation of alpha-synuclein fibril structure by site-directed spin labeling. J Biol Chem. 2007 Aug 24;282(34):24970-9. PubMed.
- Cho MK, Kim HY, Fernandez CO, Becker S, Zweckstetter M. Conserved core of amyloid fibrils of wild type and A30P mutant α-synuclein. Protein Sci. 2011 Feb;20(2):387-95. PubMed.
- El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E, Pessi A, Neill D, Wallace A. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 1998 Nov 27;440(1-2):71-5. PubMed.
- Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007 Mar 21;27(12):3338-46. PubMed.
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. PubMed.
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015 Jun 18;522(7556):340-4. Epub 2015 Jun 10 PubMed.
- Sachse C, Fändrich M, Grigorieff N. Paired beta-sheet structure of an Abeta(1-40) amyloid fibril revealed by electron microscopy. Proc Natl Acad Sci U S A. 2008 May 27;105(21):7462-6. PubMed.
- Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, Grigorieff N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):19813-8. PubMed.
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AØ, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007 May 24;447(7143):453-7. PubMed.
Further Reading
News
- More Evidence Ties Aβ Strains to Distinct Pathologies
- Watch Aβ Aggregate in Real Time Inside Cell’s Acid Vesicles
- Amyloid Fibrils Share a ‘Light’ Load
- Does Aβ Come In Strains? Glimpse Into Human Brain Suggests Yes
- Aβ Fibrils Drive Oligomer Formation, New Model Suggests
- Anti-parallel Universe—Rare Amyloid Peptides in Cylinders, Sheets
Primary Papers
- Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nannenga BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T, Eisenberg DS. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015 Sep 24;525(7570):486-90. Epub 2015 Sep 9 PubMed.
- Goedert M, Cheng Y. Parkinson's disease: Crystals of a toxic core. Nature. 2015 Sep 24;525(7570):458-9. Epub 2015 Sep 9 PubMed.
- Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fändrich M, Grigorieff N. Peptide dimer structure in an Aβ(1-42) fibril visualized with cryo-EM. Proc Natl Acad Sci U S A. 2015 Sep 22;112(38):11858-63. Epub 2015 Sep 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Max Planck Institute for Biophysical Chemistry
This highly interesting study by Eisenberg and colleagues reveals the atomic resolution structure of two short segments of α-synuclein in a fibril-like state. Using micro-electron diffraction. the structure of an 11-residue segment of α-synuclein, which is at the core of amyloid fibrils of full-length α-synuclein, has been resolved in nanocrystals down to a fascinating resolution of 1.4 Å. Together with the structure of another short segment containing the site of a genetic mutation (A53T), an interesting model for an approximate 30-residue fragment of α-synuclein in amyloid fibrils was proposed.
The proposed model goes beyond what has been previously possible on the basis of X-ray crystal structures of other short peptides, as it includes an experimentally supported turn at Gly51. The turn is important as high-resolution solid-state NMR studies of Aβ peptides have shown that amyloid fibrils of longer polypeptides are not only composed of near ideal, stacked β-sheets, but can have a variety of kinks and turns. The positioning of the NACcore at the center of the proposed model is in agreement with previous NMR studies (e.g., our work as in Cho et al., 2011), which showed that in amyloid fibrils of full-length α-synuclein the NACcore segment is most protected from hydrogen-deuterium exchange. At present, it remains open how the additional β-strands—according to solid-state NMR most polymorphs of full-length α-synuclein characterized so far contain at least four to five β strands—will be arranged in amyloid fibrils of full-length α-synuclein.
In the first solid-state NMR study of α-synuclein fibrils performed by the Baldus group (Heise et al., 2005) different polymorphs of amyloid fibrils of full-length α-synuclein were reported, and several more have been described in recent years. The high-resolution structure of the two short segments of α-synuclein in nanocrystals, as revealed now by micro-electron diffraction, does not yet provide direct information about potential differences in the structure of different polymorphs of α-synuclein fibrils. However, in the two strains of α-synuclein recently reported (Bousset et al., 2013; Peelaerts et al., 2015), the NACcore region appears to be in a β-strand conformation, such that in both these strains the model proposed by Eisenberg and colleagues might represent the core of the amyloid fibrils.
The two short segments for which the high-resolution structure of an aggregated state has now been solved by Eisenberg and colleagues are clearly two regions that are highly important for the neurotoxicity of α-synuclein. Further studies are now required to find out how well these two segments recapitulate the neurotoxic properties of full-length α-synuclein. To this end a variety of aspects should be considered. This includes differences in toxic effects such as membrane permeabilization on the one hand, and the ability to lead to efficient spreading of pathology on the other. For example, amyloid fibrils might be more efficient agents for spreading of pathology (Taschenberger et al., 2011), while intermediate aggregation states such as soluble oligomers are likely to most strongly perturb cellular membranes.
In summary, the study by Eisenberg and colleagues is an important step toward the high-resolution structure of amyloid fibrils of α-synuclein taken from different strains.
Weill Medical College of Cornell University
Rodriquez and colleagues provide the first high-resolution structure illustrating how regions of individual α-synuclein monomers contact other monomers in order to form the β-sheet-rich amyloid fibrils that are found in the characteristic Lewy bodies and neurites that are the hallmark of Parkinson’s disease and other synucleinopathies. Notably, the authors characterize the structure of microscopic crystals formed by an 11-residue segment of the NAC domain of α-synuclein, which has long been considered to contain the key elements required for synuclein aggregation (for example, by Giasson et al. and by Bodles et al.). This segment of the protein contains sequence elements that differentiate α-synuclein from its family members β- and γ-synuclein, and has been pinpointed previously.
In the crystal structure, α-synuclein residues 68-78 form a single β strand. Such a long β strand has not been previously observed in crystal structures of other amyloidogenic peptide fragments, and is posited by the authors to be responsible for the microscopic nature of the crystals that they obtained. The formation of a β strand by this particular region of α-synuclein within fibrils has been consistently documented in a number of previous solid-state NMR studies, including those from the groups of Marc Baldus, Roland Riek, Chad Rienstra, and Beat Meier, though the presence of glycine 73 in the middle of this segment has raised some questions regarding the possibility of a kink or interruption of the β-strand structure.
The crystal structure also reveals how individual copies of this segment interact with other copies to form the spine of the resulting amyloid fibrils. These highly specific interactions have proven difficult to delineate using solid-state NMR or other techniques for α-synuclein, although considerable advances have been made for other amyloids by the groups of, among others, Rob Tycko, Beat Meier, and Robert Griffin. Nevertheless, the reported structure is that of a typical twofold symmetric steric zipper, formed by two individual strands, in the characteristic manner that has now been observed by the Eisenberg group for many amyloidogenic peptides (although the presence of two water molecules within the interface is noted as an anomaly, since amyloid steric zippers typically exclude water completely). The biggest surprise and advance contained in the present work is the identification of a second interaction interface for amino acids 68-78, formed on the opposite side, i.e., the “outside” of the steric zipper, which is presumed to define the innermost core of the fibrils. Furthermore, a second synuclein fragment, consisting of residues 47-56, is found to form amyloid fibrils with an atomic interface that almost exactly mimics that formed on the “outside” of the 68-78 peptide, and features nearly identical amino acid sequences. The authors draw the obvious conclusion, namely that 47-56 of α-synuclein may fold back to pack onto the outside of the innermost fibril core structure formed by residues 68-78. This enables them to build a highly detailed model for the amyloid fibril of α-synuclein, in which the innermost core interface and the packing of a secondary layer are delineated at atomic resolution.
The new insights offered by this work have several implications for future research. First, the authors note that the A53T mutation, which was used in their constructs, likely enhances packing at the secondary interface, perhaps explaining why this mutation aggregates more efficiently and is associated with disease. It will certainly be interesting to determine and/or model the effects on this interface of the other known PD mutations in this region, H50Q, G51D and A53E. In addition, the current work does not inform about the conformation that other parts of α-synuclein may adopt in the fibril structure. On the C-terminal side, residues up to position 95 are known to be ordered within α-synuclein fibrils, and on the N-terminal side, some fibril forms exhibit order until the very N-terminus, though other forms only extend to residue 38 or so. This leaves a large portion of the fibril structure that needs to be determined and reconciled with the current model. Furthermore, the region between the 47-56 segment and the 68-78 segment is also found to adopt β-strand structure in at least some fibril forms, suggesting that this region may play a more important role than simply that of a connecting loop.
Intriguingly, recent progress in the study of several amyloids has suggested that different disease strains may correspond to different structural strains of the associated amyloid fibrils, and this hypothesis has been extended to synuclein fibrils in recent work from the Lee/Trojanowski and Melki/Baekelandt groups. Certainly, a number of morphologically distinct α-synuclein fibril forms have been characterized by different groups, and it is clear that at least some details of their molecular structures differ. It will be interesting to see whether the micro-electron diffraction technique used to obtain the current structures will be able to provide information on fibrils formed by additional α-synuclein segments and constructs, and whether such additional structures may offer deeper insights into the links between amyloid structure and disease etiology and presentation.
Drexel University
In this study, Schmidt and colleagues elucidate the Aβ42 dimer structure within the Aβ42 fibril by cryo-electron microscopy (cryo-EM). This is a follow-up from the previous study of the same group (Schmidt et al., 2009), in which cryo-EM was applied to examine possible models of Aβ40 and Aβ42 fibrillar structures that might be consistent with the experimental constraints of the technique.
Interestingly, in this earlier study two protofilaments of Aβ40 fibrillar structure were identified (one associated with a more structured N-terminal region than the other), whereas a single protofilament characterized Aβ42 fibrillar structure. The present study focuses on the Aβ42 fibrillar structure (Schmidt et al, 2015). The Aβ42 dimer unit of the protofilament consists of the central region containing two oppositely oriented C-terminal segments (each 23-26 amino acids long) forming a β-sheet structure within the parallel cross-β fibril, while the two peripheral regions formed by the two N-terminal segments (the first 10-12 amino acids in the sequence) flank the central region but are significantly less structured than it. This overall structure, based on an Aβ42 dimer unit within the fibril, differs from the Aβ42 fibril structure reported earlier this year by Ishii and collaborators, who used solid-state NMR (Xiao et al., 2015). The most obvious difference between the two is the Aβ42 tertiary fold in the decapeptide region Aβ42 (21-30) detected by the solid-state NMR, which appears to be absent from the cryo-EM fibrillar structure. This U-shaped turn structure characterizes the Aβ40 fibril model proposed by Rob Tycko and collaborators and its absence from the cryo-EM-derived Aβ42 fibrillar model is a novel and unexpected feature. Notably, the extended central region of the cryo-EM Aβ42 fibrillar structure is larger than the corresponding region of the cryo-EM Aβ40 fibrillar structure, which explains the observation that Aβ40 and Aβ42 fibrils do not mix well in vitro.
The observations of Schmidt et al. are consistent with oligomer structure predictions of our computational study (Urbanc et al., 2004), which showed that (a) the C-terminal region of Aβ42 monomer and oligomer conformations is significantly more structured than the C-terminal region of the corresponding Aβ40 conformations; (b) Aβ42 oligomerization is dominated by interactions between C-termini, whereas the central hydrophobic cluster plays the dominant role in Aβ40 oligomerization; and, most importantly, the N-termini of Aβ42 oligomers are significantly more flexible and disordered than the N-termini of Aβ40 oligomers, a feature that was hypothesized to mediate toxicity of Aβ42 oligomers (Urbanc et al., 2011; Dec 2010 conference news). Schmidt et al. posit that Aβ42 fibril is formed through self-association of dimers and/or their multiples, which would imply that some structural features of oligomers may be preserved in the fibril structure, which appears to be in agreement with computational studies. Moreover, Aβ42 fibril growth through dimer/multimer association is also consistent with the general biophysical principles of the minimal self-assembly model, recently reported by my group (Barz and Urbanc, 2014), which predicts a nucleated, structural conversion from disordered quasi-spherical to more ordered, elongated, protofibril-like structures.
References:
Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, Grigorieff N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):19813-8. PubMed.
Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat Struct Mol Biol. 2015 Jun;22(6):499-505. Epub 2015 May 4 PubMed.
Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid beta-protein folding and oligomerization. Proc Natl Acad Sci U S A. 2004 Dec 14;101(50):17345-50. PubMed.
Urbanc B, Betnel M, Cruz L, Li H, Fradinger EA, Monien BH, Bitan G. Structural basis for Aβ1–42 toxicity inhibition by Aβ C-terminal fragments: discrete molecular dynamics study. J Mol Biol. 2011 Jul 8;410(2):316-28. PubMed.
Barz B, Urbanc B. Minimal model of self-assembly: emergence of diversity and complexity. J Phys Chem B. 2014 Apr 10;118(14):3761-70. Epub 2014 Mar 6 PubMed.
Make a Comment
To make a comment you must login or register.