Held in a historic skyscraper built in 1932 for a Philadelphia bank, the seventh conference on Clinical Trials on Alzheimer’s Disease drew 715 scientists to this city between November 20 and 22. CTAD featured a sprinkling of new trial results and enthusiasm about treating agitation in AD, but most of the activity reflected a field trying to rebuild itself from the ground up. Trialists swapped notes on implementing new diagnostic criteria in therapy trials, enriching trial populations, and exploring home-based assessments and other tools to support prevention trials. Secondary prevention sounded positively mainstream and has become the stuff of large-scale collaborations. Rusty Katz, formerly of the FDA, implored trialists to stop obsessing over disease modification and to aggressively go after big therapeutic effects instead. Those, Katz said, may require a commitment to co-develop combinations of investigational drugs.
CTAD Shows Alzheimer’s Field Trying to Reinvent Itself
When 715 scientists gathered in Philadelphia for the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, their presentations and debates reflected a field that is revamping itself in hopes of more success in the next wave of drug trials. For the past decade, clinician-researchers have discussed innovation and laid the groundwork; however, in practice most intervention trials still hewed to the conventional formula of parallel group studies in clinically diagnosed mild to moderate Alzheimer’s, with little use of biomarkers. Now, all that is changing. “Things are completely different from five years ago in the drug development process,” said CTAD co-organizer Paul Aisen of the University of California, San Diego, who heads the Alzheimer’s Disease Cooperative Study (ADCS).
CTAD took place in the PSFS Building. Built in 1932, this T-shaped banking tower is sometimes considered the “first international-style skyscraper” of the United States. A National Historic Landmark, it is now a Loews hotel.
Even as change is unfolding, however, it is too timid for some. The best-received talk at CTAD was a keynote by Rusty Katz, who directed the FDA’s neurology division until last year. Katz exhorted the field to stop spinning its wheels trying to earn a label of disease modification. Rather, trialists should focus on delivering a large therapeutic effect, regardless of whether the drug changes progression or symptoms. That would honor the therapy goals of the National Alzheimer’s Plan, he said, and may require co-developing several investigational drugs together (see Part 2 of this series).
By now, researchers across the field fully recognize the importance of the roughly 15-year preclinical phase of Alzheimer’s. “DIAN [the Dominantly Inherited Alzheimer Network] and API [Alzheimer’s Prevention Initiative] have shown us that there is a sequence of biomarker appearance, and AIBL [the Australian Imaging, Biomarkers and Lifestyle study] has helped us have a lot of faith in this sequence,” Rachelle Doody of Baylor College of Medicine in Houston said in her keynote address. Prospective studies are largely convergent in their finding that brain amyloid predicts cognitive decline, albeit at rates that are subject to other risk factors, as well (see part 11 of this series.) Trialists have begun to harness this long preclinical window of time for therapy development. This means new ways of doing things across all steps of the process. Secondary prevention trials—in people who have evidence of AD biomarkers but no or very subtle symptoms—have begun enrolling based on modern diagnostic criteria, and their leaders are swapping notes on what needs tweaking as first experiences roll in. Leaders are banding together in groups such as the Collaboration for Alzheimer’s Prevention (CAP) to coordinate their searches for outcome measures and biomarker procedures for such trials. The DIAN, API, and Anti-Amyloid Treatment in Asymptomatic Alzheimer's Disease (A4) trial platforms have inspired the European Prevention of Alzheimer’s Project (EPAD), an international consortium to build a standing platform of secondary prevention trials (see Part 3 of this series). Registries are springing up in the United States and Europe to help these large studies recruit asymptomatic participants. “The idea of secondary prevention has become mainstream thinking,” said Aisen.
Even as the first secondary prevention trials are already enrolling, it is clear that some of the newer techniques they are using need refinement as the trials proceed. For example, a Phase 2 trial of Genentech’s therapeutic antibody crenezumab showed that amyloid PET may require a different reference region than used before—and than specified in current prevention trial protocols—if PET is to reliably gauge a drug effect in longitudinal measurements (see Part 4 and Part 5 of this series). Scientists know that using evolving methods in trials poses risks, but they have to do so anyway because following tried-and-true methodology has led them down dead ends for the past 20 years.
Despite being a truly nascent technique, tau PET is expected to be included in secondary prevention trials as soon as January 2015. “Tau PET is clearly a game changer,” Aisen said. Ask any trialist, and they express equal excitement about being able to both image defining Alzheimer pathologies in living people and to test more tau-based drugs (see tau therapeutics). As researchers scan observational cohorts and compare results against Braak staging of tau pathology, a unifying idea is rapidly gaining currency: that some tau pathology accumulates in most people’s medial temporal lobe as they age, but in the large subset of older people who accumulate amyloid, the tau pathology changes from a circumscribed process of aging to a propagating lesion that claims synapses and brings on AD symptoms (for more on tau, see Part 6 of this series.)
Researchers are stacking their initial secondary prevention trials with exploratory cognitive measures in hopes of moving beyond the paper-and-pencil tests that trial volunteers have been taking for decades. Home-based iPad tests and dementia staging tools, as well as patient-reported outcomes, already are being validated to generate richer datasets even while relieving study volunteers of the burden of having to trek to their memory clinic as often (see Part 7 of this story).
The next frontier, in preparation for primary prevention trials, will be mining large-scale, Web-based gaming data or unsupervised cognitive assessments taken by tens of thousands of people. This could help clinicians spot subtle decline brought on by soluble forms of amyloid. “Remember, we do not think fibrillar amyloid is the major driver in Alzheimer’s disease,” said Aisen. “If that is true, then we should be able to find a cognitive indicator of soluble amyloid toxicity prior to big masses of insoluble deposits spreading across brain and fueling big masses of tau spreading.”
That is the future, however. The present, on display at CTAD, features innovation in early stage trials—innovation that was guided in part by recent revisions of the diagnostic criteria for Alzheimer’s disease. Leading an international group of clinicians, Bruno Dubois of the Pitié-Salpétrière Hospital in Paris first introduced new criteria in 2007, then proposed a new vocabulary for what to call the early stages in 2010. “These attempts at nosology have been very helpful at stimulating a thorough discussion,” Doody said. This past summer, Dubois and the international working group further updated the criteria in light of longitudinal biomarker data gathered since 2010 (see Dubois et al., 2014).
At CTAD, Dubois said that the new criteria use a single framework for the range of preclinical and symptomatic AD. They require biomarkers as part of the diagnosis at each stage. The criteria split the biomarkers such that only the CSF signature and amyloid PET remain part of the diagnosis. In essence, the pathophysiological markers are integral to the Alzheimer’s diagnosis, but volumetric MRI and FDG PET got kicked out. Why? The former are specific to AD, the latter are not, hence they are less useful for diagnosis. Rather, volumetric MRI and FDG PET are suited for staging and monitoring progression because they stay dynamic as disease progresses, Dubois said.
The new Dubois diagnosis always has two components. The biomarker component requires either the pathological CSF signature of both Aβ/tau or amyloid PET (or an autosomal-dominant mutation), while the clinical component varies with the person’s presentation. For example, a diagnosis of typical AD requires at least an episodic memory impairment—isolated or with other cognitive or behavioral changes—that got gradually worse, plus the biomarker component. A diagnosis of atypical AD requires a clinical phenotype of either the posterior, logopenic, frontal, or Down’s syndrome variant of AD, plus the same biomarker component. A diagnosis of mixed AD requires an AD clinical phenotype with pathology biomarker to establish that AD contributes to the mixed disorder, plus clinical and biomarker evidence of either cerebrovascular or Lewy body disease.
For the diagnosis of preclinical AD, Dubois et al. maintain a distinction that came into question at CTAD. Among aging people with biomarker evidence but no clinical phenotype, Dubois gives a diagnosis of “presymptomatic” AD only to those few who carry a pathogenic mutation in APP, presenilin, or have the Down’s syndrome APP triplication. The much larger group of cognitively normal people who have the AD biomarker component but no such gene are told they are “asymptomatic-at-risk” for AD. Dubois acknowledged that data from longitudinal studies is increasingly suggesting that these people have AD and will develop symptoms, but so far he declines to disclose this to individual patients so long as he cannot predict when a given person will progress.
Dubois conceded a point of debate between him and other clinicians. He acknowledged that other episodic memory tests besides the free and cued selective reminding test (FCSRT) also detect the amnestic hippocampal deficit that anchors the clinical part of the AD diagnosis. In addition, the FCSRT may not be sufficiently specific when used by itself. This became clear when Philip Scheltens of Vrije University Medical Center in Amsterdam presented the way screening and enrollment had played out for Roche’s Phase 3 SCarlet RoAD trial of the therapeutic antibody gantenerumab. This trial uses the 2007 Dubois criteria. Among 2,290 potential trial participants who failed screening were 621 who met the FCSRT cutoff but did not have AD biomarkers.
Overall, these criteria drew wide praise for their simplicity. For example, Steve Ferris of New York University said, “I applaud the evolution of these criteria.” “They are very well done,” agreed Charles DeCarli of the University of California, Irvine. Three sets of revised diagnostic criteria have been developed independently in the United States, as well, and are broadly similar.
Even so, the question of how best to apply new diagnostic criteria in clinical trials is far from settled. Different trials are experimenting with slightly different ways of creating groups. Drawing an analogy with infectious disease, Doody suggested that clinical trials might simply divide people into categories of “At Risk,” and define the risk, and “AD,” and define the severity (see image).
At CTAD, Rachelle Doody cited the classification of the Ebola virus to make her point that clinical trials in Alzheimer's could use a simpler approach to creating study populations. [Images courtesy of R. Doody.]
“The most exciting progress we are now implementing in trials is thanks, to a large extent, to new diagnostic criteria,” said Niels Andreasen, who oversees clinical trials at the Karolinska Institute in Stockholm. These research criteria are used primarily in trials and expert settings. For them to become widely used, lumbar punctures or PET scans would need to become more available, and both biomarker measures and memory tests need robust cutoffs. Countries differ in how quickly they embrace the new criteria. In Sweden, many doctors already do, and they offer current AD drugs to barely symptomatic people. “I believe it is unethical to start a patient with biomarkers but no dementia on the placebo arm of a therapeutic trial without offering currently available medicines. If I know from my assessment that this is AD, even if the MMSE [Mini Mental State Examination score] is 28, I want to offer those medications,” said Andreasen.
On the other hand, some clinicians prefer not to subjugate their clinical impression to a biomarker result. For example, the Dubois criteria require either amyloid PET or CSF Aβ/tau positivity for an AD diagnosis, and the regulatory label for amyloid PET tracers states that a negative scan rules out a diagnosis of Alzheimer’s. Even so, in a study at memory clinics in northern Italy, most dementia specialists resisted changing a clinical AD diagnosis they had made in the face of a subsequent negative amyloid PET scan, Giovanni Frisoni of the University of Geneva reported at CTAD. “Dementia experts still rely on the clinical phenotype, even when amyloid PET tells them otherwise,” Frisoni said. Others later quipped that clinicians do not like being shown that they were wrong, but that this will change in the next few years once amyloid and tau PET become more established.
While secondary prevention dominates the discourse these days, there is fresh energy in other areas, too. CTAD showcased a new push to develop hormone-based therapies based on women’s health research (see Part 8 of this series). There was broad excitement about a flurry of trials to try to calm agitation and aggression in Alzheimer’s and even Down’s syndrome (see Part 10 of this series). “Nothing is more important in symptomatic AD than managing the behavioral abnormalities. If this is true, it is big news,” said Aisen.
As new trials get going, studies in symptomatic patients would be well advised to take into account a feature of Alzheimer’s that can stump any longitudinal study, namely that AD progresses faster in some people than others. Both intrinsic and external factors can influence how quickly the disease marches on in a given person. However, new trials could account for different progression rates, said Doody. She suggested collecting the National Adult Reading Test (AMNART) at screening as a surrogate of cognitive reserve and information on background medications, both of which can slow a person’s progression. Parkinsonism and psychosis should be documented, as they can speed up progression. Slow and fast progressors could be balanced across treatment arms, and pre-progression indicators incorporated in the analysis, Doody said.—Gabrielle Strobel
Rusty Unleashed: Forget Disease Modification, Go for Big Effect
In current affairs, sometimes the recently retired general gives the best advice. He speaks from experience and from the heart, no longer toeing the party line. Researchers at the Clinical Trials in Alzheimer’s Disease (CTAD) conference, held November 20 to 22 in Philadelphia, got their version of frank talk from Rusty Katz. In 2013, Katz retired—to widespread regret—from his post as director of the Food and Drug Agency’s Division of Neurology Products after 30 years with the agency. Having facilitated drug evaluation at earlier disease stages, Katz promised to stay engaged. And he does.
At CTAD, Katz urged the field to stop obsessing over disease modification, echoing a sentiment expressed earlier that day by Rachelle Doody of Baylor College of Medicine in Houston (see Doody 2008). Instead, people should focus squarely on achieving a large therapeutic effect. This, Katz said, is the way to honor the Obama administration’s National Plan to Address Alzheimer’s Disease. Where to find those big effects? Try combination trials, Katz said. He challenged the field not to hide behind regulatory obstacles. FDA guidance for this exists, waiting for trialists to rise to the task.
Katz’s keynote garnered rousing applause. Subsequent speakers and hallway commentators alike praised it. Will it change the conversation? Below, detailed excerpts from his talk.—Gabrielle Strobel
Rusty Katz at CTAD 2014:
“I want to talk to you about an approach I believe the field should pursue aggressively. I encourage you to consider adopting it as a field and committing yourselves to it now or in the very near future.
“In 2012, the Obama administration announced a national plan to combat Alzheimer’s disease. Do we talk about this anymore? The plan is ambitious. The first of its five goals is the development of an effective prevention or treatment for AD by 2025. That is not far in the future. It is a serious plan, a serious goal. We ought to keep it in mind.
“The plan calls for effective treatment. Well, at least according to the regulatory definition, we already have effective treatments. They knew that when they wrote the plan. Obviously, the plan calls for something more, something much bigger. It seems to me the plan anticipates that by 2025 we will have treatments that have large, important effects. Think about it. The treatments we are currently developing hardly fall into that category.
“Nowadays, everybody talks about treatments designed to slow disease progression. I think this is a psychological problem. It is a conversation stopper. The Holy Grail is disease modification, and we think once we do that we are more or less done.
“I think we conflate disease modification with a very large, important effect. But of course these are completely different things: You can have an effect on disease progression that is very small. That, I would submit, is not what the formulators of the national plan had in mind.
“I agree when Rachelle Doody says we ought not to think so much about disease progression. It really is a distraction. Why?
“Disease modification is very difficult to establish. I spent years at the FDA trying to answer the question from everybody: ‘How do we demonstrate disease modification?’ Quite frankly, we never gave a truly satisfying answer because we do not have a truly satisfying answer.
“We said the agency will consider an effect on a clinical outcome and a biomarker as possibly demonstrating an effect on disease progression. You ask: ‘Which biomarker?’ We say: ‘We do not know. Just measure a lot of them and at the end we will sort it out.’ This reflected the state of the science at the time, but it violates our principle of prospectively designating primary outcomes.
“We also recommended using randomized start or withdrawal designs, but these studies are very complicated and entail numerous assumptions that might not turn out to be true.
“We do not know how to establish disease progression with certainty. It is quite possible that you might develop a drug that is disease-modifying but you can’t establish that to the agency’s satisfaction. There are many things that are true but that can’t be established for certain.
“Getting a claim for disease progression is very difficult. Why all the effort to design a study that will come up with evidence about modifying progression that the agency may or may not find convincing? Why bother putting that package together?
“It is only a labeling issue. I put ‘only’ in quotation marks because I understand it’s a big deal to companies. But it really is only a labeling issue. Disease modification is not an approval issue.
“We all want approved treatments that have big effects. That is where the focus should be.
“One thing to consider [is] the multiple pathologies beyond amyloid and tau that are in the Alzheimer's brain early on. They are being talked about a lot at numerous meetings. Many are intrinsic to Alzheimer’s and related to amyloid and tau, but there are also comorbidities, for example hypertension, diabetes, inflammatory disease, Lewy bodies, hippocampal sclerosis, TDP43 deposits, vascular disease.
“The existence of these pathologies and comorbidities argues for a multi-target approach. In my view and in the view of many people, this clearly argues for developing combination therapies.
“The guidance says such combinations are appropriate when there is a serious condition, a strong biological rationale for combining therapies, when a nonclinical or short clinical study suggests that the combination is superior to the available therapy. Also when there is a compelling reason why the individual components cannot be developed independently. It is easy to make the case that all these criteria are met for Alzheimer’s disease. That is not controversial at all.
“The two big issues in co-developing investigational drugs are the non-clinical and the clinical requirements. Let’s look at them. The non-clinical safety studies for combinations are handled in Guidance M3(R2). It talks about early stage and late-stage entities; the former don’t have much clinical data, the latter do.
“The guidance talks about what to do when combining late- and early stage entities together, but there’s little clinical experience on the combination. In this situation, one-month clinical studies are appropriate, but longer clinical trials would require a toxicity study of the combination. It talks about late-stage entities when there is adequate clinical experience with each component and some experience with the combination. In this situation, you generally need no new animal studies. If there isn’t adequate clinical experience with the late-stage entities together, then you would need an animal combination study before a large Phase 3. If you have early stage entities with little or no clinical experience, then you need not complete non-clinical development programs for each individual one. A non-clinical toxicology program with the combination alone can be appropriate.
“For the clinical studies, you have to show that each component makes a contribution to the effect of the combination. This is a reasonable premise to ensure people do not take unnecessary drugs. In Phase 1 you should do something for each component individually, in Phase 2 you can attempt proof of concept for the combination. The guidance asks that you ‘further demonstrate the contribution of each drug to the extent possible and needed, i.e., the extent not sufficiently established by existing data.’ How do you show a contribution of each component? The guidance does not require any set level of statistical evidence, it just says you ought to attempt proof of concept in Phase 2.
“Typically people use a factorial design for combinations of two drugs, i.e., AB vs. A vs. B. For a combination of three or four components it is unclear what a factorial design would look like, but it is likely to be large and complicated. So the question is: Is it necessary? Could it be shown in animals instead? I do not know. But the guidance says: ‘Full factorial studies in Phase 3 need not always be performed.’ The critical point in the guidance may be this: ‘If findings from in vivo or in vitro models and/or a Phase 2 trial adequately demonstrate the contribution of each new investigational drug to the combination, Phase 3 trials comparing the full combination to a control or placebo generally will be sufficient to establish effectiveness.’
“At least to me, this means that animal or in vitro data may be able to demonstrate the contribution of each component, hence Phase 3 really does not have to be terribly complicated. This is very much worth keeping in mind.
“There has been considerable reluctance to doing combination studies. Arguments I have heard include cost, risk, poor correlation between animal and human. Yes, but that is true in most diseases. We deal with this all the time. Target validation is a big question. How do you know you have engaged the target? That, too, is true in all sorts of indications.
“Intellectual property questions are cited. How do you share data with other companies if you develop a combination together? You just need to think through these problems and solve them.
“Some of you believe you need to fully develop the individual components first. Please be clear that that is not necessary. There are paths to develop two, three, or four investigational drugs together from Day One. Some problems I hear cited are real, others are just misperceptions, but the important thing is: they all can be dealt with.
“One way to approach combinations is with adaptive clinical trial designs. The agency in 2010 issued a draft guidance on this (see FDA Guidance). Adaptive designs have to be planned well and they are complex. However, from a clinical perspective they are more efficient designs. This guidance tells you that almost all aspects of a trial can be adapted: from eligibility criteria, randomization, treatment regimens and arms, sample size, even to some endpoints.
“The main worry with adaptive designs is the danger of inflation of type-one error, and the document is mainly concerned with avoiding that. This is a big area I cannot discuss fully, but I encourage you to use adaptive designs for exploratory studies, Phase 1 and 2.
“One example of an adaptive design worth highlighting is the futility study. Its purpose is to figure out early that your treatment does not work. That’s the goal. You do this early in Phase 2. You set a treatment effect that you want to see and you declare futility if your treatment does not reach that effect size. This is critical for the point I am trying to make: that we are looking for a large effect.
“A futility study helps you figure out early if your drug or your combination is going to have a big effect. You can do this in one trial with different combinations of drugs A, B, and C. This is very important: Remember we are looking for large effects.
“At this point in time, we should focus on large effects regardless of whether they are disease-modifying or—more accurately—whether we can tell if they are. Whether a treatment is disease-modifying or not is not particularly important to patients. Of course, the agency will continue to approve treatments with small effects. The agency typically does; I spent 30 years doing that. But think about it: Is it too radical to suggest that maybe we should pass up those treatments? That we should design trials to get rid of small effects early and deliberately go after large effects?
“I hope you see that FDA regulations permit the development of combination therapy regardless of the development stage of the components. Paths are laid out to help people do that.
“Adaptive design at all phases can be helpful in expediting the approval of combinations. But let’s not fool ourselves: These studies are complicated. They require a lot of planning, and there is little neurology experience with them in the field and at the FDA. People in other fields have a lot experience, however, and I encourage you to engage those people.
“It makes sense to start planning an infrastructure now. A group could decide what combinations to study, begin to design trials, and involve multiple companies and the FDA in this early effort. I would hope the agency is interested in collaborating with that. Especially in the design of Phase 2 studies to establish the contribution of each component, you would want their input early on.
“The clock is ticking. There’s a plan out there. Those of us who were alive in the ’60s remember President Kennedy’s 1961 goal of reaching the moon by the end of the decade. We have a few more years to reach our goal than they had. They were only dealing with the laws of physics and we are dealing with the human brain, which is more complicated, to be sure. But nonetheless, they focused on their goal, and they met it.”
From Shared CAP, Secondary Prevention Trials Are Off and Running
In 2011, as the Dominantly Inherited Alzheimer Network (DIAN), Alzheimer’s Prevention Initiative (API), and Anti-Amyloid Treatment in Asymptomatic Alzheimer's Disease (A4) trial platforms were each moving into place, their respective leaders realized they had much to gain from working together, and they formed an umbrella group called Collaboration for Alzheimer’s Prevention. CAP is a forum to coordinate development of new science needed for secondary prevention trials. Its scientists collaboratively try to ensure that its constituent trials gather data in a way that allows direct comparison, and seek public-private funding that facilitates sharing of data and samples with the field at large (see Aug 2012 news story). Having met regularly, this year CAP hosted the opening symposium at the 7th Clinical Trials on Alzheimer’s Research conference, held November 20 to 22 in Philadelphia, with an update on where their projects stand.
The Gordian knot of secondary prevention is to show a clinical benefit in people who have no symptoms. The many facets of the problem—from defining and recruiting trial populations to reducing measurement variability of biomarkers and creating outcome measures that will satisfy regulators—make it too big for an individual academic or industry group to solve. CAP reflects a realization that cutting the knot will require cooperation among many groups. “Secondary prevention does not take a village, it takes a federation,” quipped Pierre Tariot of the Banner Alzheimer’s center in Phoenix, the U.S. home of the API trials platform.
Besides leading clinicians of the DIAN, API, and A4 trials, the CAP umbrella includes Duke University’s Kathleen Welsh-Bohmer of the TOMMORROW study, as well as representatives of the National institute on Aging, the Food and Drug Administration, the Alzheimer’s Disease Cooperative Study, the Alzheimer’s Association, and the Fidelity Biosciences Research Initiative. (Fidelity provides financial support to Alzforum.) Each trial platform represents a large group of collaborators.
At CTAD, Randall Bateman of Washington University, St. Louis, cited as an example of CAP’s work its members' development of a cognitive outcome measure for their respective secondary prevention trials in close coordination with one another. New outcomes were needed because the CAP trials were enrolling asymptomatic or very mildly symptomatic patients in whom established cognitive batteries for mild to moderate Alzheimer’s disease were insensitive. This effort started with an API project led by Jessica Langbaum at the Banner Alzheimer’s center to analyze data from different longitudinal aging and AD cohorts in search of the handful of tests that changed the most during a person’s preclinical decade, but varied least from one person to the next. Soon after, Reisa Sperling of Harvard Medical School and Mike Donohue of the University of California, San Diego, started a similar project for A4, and Jason Hassenstab at Washington University St. Louis, Peter Snyder from Brown University, in Providence, Rhode Island, and others did one for DIAN. Each group, using different approaches in different datasets, arrived at largely the same composite of five cognitive tests that they are now using in their respective trials. At CTAD, Sperling said that each composite uses slightly different individual tests; the important thing is that each composite taps with equal weight into the same domains of cognition, such as episodic memory, attention, and executive function.
In CAP, scientists exchanged notes frequently on their respective efforts at devising new cognitive outcome measures for secondary prevention trials. [Image courtesy of R. Bateman, DIAN-TU.]
Moreover, the trial platforms jointly evaluate cognitive tests on iPads in hopes of gradually conducting more and more aspects of a prevention trial in people’s homes. Working with Dorene Rentz at Brigham and Women’s Hospital in Boston, the A4 group developed an iPad cognitive composite that contains some tests from the commercial CogState battery. They added a face-name task the Boston group has studied for years in preclinical populations using functional MRI, as well as a pattern-separation task that is sensitive to early hippocampal changes. The trials unit of DIAN (DIAN-TU) uses a similar iPad composite. DIAN-TU went a step further in transitioning parts of the trial into the home by hiring and training home health nurses near where trial participants live to visit and deliver study medication or placebo every month.
The ongoing DIAN, API, and A4 trials are all heavily loaded with biomarkers. CAP members have centralized data collection procedures for spinal fluid and imaging biomarkers. They standardized and shared protocols, as well as methods of analysis, Bateman said. “This is a huge deal to all of us,” said Tariot. “If we can show that a biomarker effect in two years powerfully relates to a clinical benefit in five years, then that opens the door for the whole field to conduct more efficient and shorter biomarker studies.”
All original CAP members have pursued public-private funding for their trials as a way to ensure that trial data, and even some samples, can be more readily shared across the scientific community than is typically the case with industry studies. National Institutes of Health and philanthropic grants frequently require that data and sample aliquots or scans be made available for other studies. The CAP members seek permission for this kind of sharing in the consent form they use when they enroll participants. They have worked out a process to release data in stages. For example, A4 screening data—including thousands of amyloid and tau PET scans—will become available when that study is fully enrolled, longitudinal data will become available when the study is complete, and treatment data when the regulatory process is complete, said Sperling. In the DIAN-TU trial, the sponsoring drug companies and the public-private DIAN-TU co-own the data and samples.
Some instruments, such as the cognitive composites, are already available. Another advantage of the public-private arrangement is that it can make a trial stretch toward perhaps-not-quite-achievable research goals, said Sperling. For example, A4 has mandated that 20 percent of screens be done in potential participants from an underrepresented minority. “This may be impossible, but an academic-private partnership allows us to try to understand what factors influence risk in African-American and Latino populations,” Sperling said.
Coordination notwithstanding, the trials differ in important aspects. For example, DIAN-TU, which enrolls people with autosomal-dominant APP or presenilin mutations across three different continents, has to the make the most of small participant numbers. It tries to maximize power from its current enrollment target of 210 people by using an adaptive design that tests gantenerumab and solanezumab against a pooled placebo group. The API’s Colombian trial, in 300 relatives of a kindred afflicted with the E280 presenilin (Paisa) mutation, is a single-center trial conducted in Medellin, Colombia, with a handful of satellite sites so people travel shorter distances for twice-monthly injections and safety checks. Both trials conceal mutation status, and so must include some non-carriers, all of whom are randomized to placebo. Both enroll easily, however, from an existing observational cohort of deeply phenotyped and motivated family members, hence almost no candidates fail their screen. DIAN-TU thus far has randomized 52 people at 20 sites, with one screen failure and no dropouts, Bateman said at CTAD.
In contrast, A4 aims to enroll 1,100 people with biomarker evidence of brain amyloid at 60 centers across North America and in Melbourne, Australia. The luxury of a much larger cohort allows this trial to be simpler in one way, said Paul Aisen of the University of California, San Diego. A4 is a straight-up parallel-group trial comparing one drug, solanezumab, to placebo. This avoids the potential statistical pitfalls that make some trialists nervous about adaptive designs until there is some experience with them in neurologic diseases. Rather, the challenge in A4 is to muscle through the sheer effort of screening. A4 needs to screen up to 10 people for each one it enrolls—yeoman’s work for both the sites and the potential participants.
Why so many screen failures? Sperling said a disproportionate number of people scored below the cognitive cutoff set in the protocol, largely for lack of appropriate adjustment to educational background. To address this, in one of several tweaks to the screening parameters, A4 broadened the range of cognition to allow more older people with a family history, an ApoE4 allele, and perhaps subjective memory concerns to become eligible for a subsequent screening PET scan, Sperling said. Other trials, too, are feeling their way toward the right cognitive and biomarker cutoffs as they screen pre-dementia cohorts.
At CTAD, Sperling said that to date A4 has screened 631 people. Thus far, 36 have received study medication and additional participants are eligible to be randomized. The first participant infused in June, in Providence, Rhode Island, allowed his infusion to be photographed by the Associated Press. “Some A4 participants have buried their parents with AD and have an older sibling in a nursing home. They are cognitively normal but are willing to go public and say, ‘I have amyloid and want to do something about it,’” said Sperling.
In 2013, Welsh-Bohmer joined the CAP group. She is the neuropsychology lead for the TOMMORROW trial of low-dose pioglitazone, which also enrolls participants at a pre-dementia stage of Alzheimer’s disease. Beyond that, this trial is different in many ways. Funded entirely by the companies Takeda and Zinfandel Pharmaceuticals, there is no public-dollar leverage to ensure early sharing of all data or samples.
Unlike DIAN, API, and A4—which ascertain amyloid positivity in their participants and have chosen investigational anti-amyloid drugs for their first trials—TOMMORROW is testing whether an approved diabetes drug can stave off a diagnosis of mild cognitive impairment due to AD. The study is based on the bioenergetics hypothesis of Alzheimer’s disease and posits that targeting mitochondrial, inflammatory, and metabolic abnormalities in Alzheimer’s pathogenesis with pioglitazone will delay progression. In addition, the trial aims to qualify a genetic biomarker risk algorithm developed from Allen Roses’ proposal of a TOMM40 length polymorphism (Roses et al., 2010). The trial splits cognitively normal people into a low-risk group and a high-risk group, treats half of the latter with 0.8 mg of pioglitazone daily, and then measures time to diagnosis of MCI due to AD in all three groups, Welsh-Bohmer said.
TOMMORROW is the largest trial in CAP. It aims to enroll 5,800 people between the ages of 65 and 83—4,600 for the high-risk group—at 60 sites across the United States, Europe, Russia, and Australia. This trial uses neuropsychology criteria for a diagnosis of MCI due to Alzheimer’s (Alberts et al., 2011) as its primary endpoint. Different sites in different countries implement MCI criteria differently, and the cognitive tests used in the trial have not been culturally validated; nor are preset normative data available for non-English countries such as Russia, Italy, Germany, or Switzerland. Therefore, the TOMMORROW team first conducted a validation and norming study. This pre-study has concluded enrollment, and sites in those countries will launch the actual trial in 2015, Welsh-Bohmer said. In English-speaking countries, the trial started in August 2013. Thus far, 37 sites have randomized some 1,000 patients with a screen failure rate of seven to one, she added. Welsh-Bohmer said that the TOMMORROW trial adds diversity to the CAP in its approach to risk stratification, the clinical endpoint selected for determining efficacy, and treatment.
Last but not least, while these four secondary prevention initiatives are out of the gate, a fifth is gearing up for a formal start in January 2015. At CTAD, Craig Ritchie of the University of Edinburgh, Scotland, U.K., told the audience that on November 14 the European Innovative Medicines Initiative (IMI) approved a handsome €64 million toward the European Prevention of Alzheimer’s Project (EPAD). EPAD is a public-private consortium to create a platform for a standing proof-of-concept trial of 1,500 people for the secondary prevention of AD. The idea is to gather some 24,000 people in a registry and enroll 6,000 of them into a longitudinal cohort of cognitively normal people who will be characterized with biomarkers and cognitive measures broadly similar to what API and DIAN-TU have been doing with their respective cohorts. Ritchie said that this biomarker phenotyping would be done to industry standards and create a reservoir population for intervention trials of people with biomarker evidence of AD who have very mild cognitive symptoms or none at all. The trials would evaluate proof of concept against a biomarker outcome of a succession of investigational drugs in multiple combinations of arms, emulating the principle of the I-SPY-2 trials platform for cancer.
Using an adaptive design, EPAD trials are to proceed in two steps. Any given drug is first evaluated against an intermediate biomarker that is linked to the mechanism of the drug—amyloid for an anti-amyloid drug, tau for an anti-tau drug. If it succeeds, the drug will go on to a second evaluation against a cognitive/clinical outcome. This outcome does not change from drug to drug and supports regulatory approval.
Even with €64 million, the project is so large that it cannot start from scratch, Ritchie said. He is reaching out to investigators of pre-existing aging cohort studies across Europe to invite them to collaborate. Ritchie called the amount of collaboration needed for EPAD to succeed “staggering.” He showed organizational charts and work package descriptions for how 35 partner organizations in academia and industry are going to work together. To pull the project off, EPAD will establish approximately 30 so-called trial delivery centers across Europe, starting with centers in Edinburgh, Scotland; Toulouse, France; Stockholm; and other cities. EPAD will formally kick off at a meeting on January 14 in Paris. —Gabrielle Strobel
Many researchers are eagerly watching for results from Alzheimer’s disease immunotherapy trials. The 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20 to 22 in Philadelphia, brought a smattering of updates on these programs, although no big news. Several talks highlighted methodological problems and how researchers are tweaking trial protocols to better measure outcomes and select participants. Biomarker data from the Phase 2 trial of Genentech’s crenezumab reinforced concerns about the standard method of analyzing amyloid PET data, and speakers argued that using a white-matter reference region could help them better detect how the amyloid burden changes over time. Regarding recruitment, other talks stressed the importance of screening for brain amyloid, as it appears that many people who meet cognitive criteria for prodromal AD do not have it. Beyond these technical issues, there were updates on antibody trials, including mostly negative results from a couple of active immunotherapy approaches that have now been scuttled (see Part 5 of this series).
First, consider crenezumab, Genentech’s antibody that binds all forms of Aβ. The Phase 2 results in the mild to moderate ABBY trial were negative overall, with hints of a benefit in patients with milder disease (see Jul 2014 conference news). At CTAD, Stephen Salloway of Butler Hospital, Providence, Rhode Island, presented biomarker data from the Phase 2 BLAZE trial. In this study, 91 participants with mild to moderate AD received either placebo, 15 mg/kg crenezumab injected intravenously, or 300 mg/kg crenezumab subcutaneously for 73 weeks.
On the primary endpoint—PET amyloid imaging—the researchers saw no consistent trend and no difference between the treatment groups on the prespecified analysis. Salloway noted that results from the three scans each patient underwent varied widely, and more than expected. Paul Aisen of the University of California, San Diego, who was not involved with this study, said that Alzheimer’s trials in general still have several biomarker measurement problems that need to be worked out. On the other hand, the secondary endpoint in the BLAZE trial, CSF Aβ, rose slightly but significantly in both treatment groups, in contrast to continued decline in the placebo group, Salloway reported.
On the PET measurement issue, imaging experts have been arguing for some years that the standard method of assessing amyloid PET changes against cerebellar gray matter as a reference region introduces too much noise into the data, especially for longitudinal measurements. White matter, which takes up less tracer, may provide a better contrast (see Feb 2012 conference news). For this reason, Genentech authorized two secondary analyses using white-matter reference regions. Eric Reiman at Banner Alzheimer’s Institute, Phoenix, presented the results from a blinded analysis using a cerebral white-matter reference region that lies at roughly the same plane of the scanner “slice” as the amyloid. In ADNI longitudinal data, this reference region lowers variability and more faithfully reflects the difference between amyloid progressors and non-progressors (see Jul 2014 conference story). With this method, trajectories of amyloid deposition in the crenezumab BLAZE cohorts looked smoother and had less variability, Reiman reported at CTAD. A treatment effect emerged in the IV group, where amyloid appeared to accumulate more slowly, although the effect missed significance. “I believe we are underestimating the treatment effect with crenezumab,” Reiman said.
Gilles Tamagnan at Molecular NeuroImaging, New Haven, Connecticut, led the second exploratory analysis. It used white matter in cerebellum and other subcortical regions as a reference. Salloway discussed this data at CTAD, noting that the findings looked similar to Reiman’s, with the IV treatment group trending toward less amyloid accumulation. As with Reiman’s method, the within-person variability of longitudinal scans dropped.
Spatial distribution of amyloid PET tracer binding in Alzheimer's disease. [Image courtesy of Felix Carbonell, Biospective Inc.]
Which method is better? Barry Bedell of McGill University, Montreal, addressed this question in his talk. In an analysis of PET data obtained from ADNI cohorts and participants in Biogen Idec’s Phase 1b trial of the Aβ antibody BIIB0037, white matter in the cerebellum and pons provided the best reference region for discriminating amyloid-negative from amyloid-positive scans, he reported. Bedell told Alzforum that cerebral white matter performed comparably to cerebellar white, but during immunotherapy the former tissue can develop lesions known as ARIA-E, which affect the PET signal. For this reason, a subcortical reference region may be more reliable. However, Bedell stressed that the key was to exclude cerebellar gray matter. “Every time we included cerebellar gray matter, the results were inferior to when we did not. There is a marked increase in accuracy when you exclude it.”
In the crenezumab trial, the hint of a treatment effect occurred only in the IV groups. In the subcutaneous treatment group, which received about half the effective dose as the IV group because not all the injected antibody got into the bloodstream, trajectories resembled those of controls. As a result of these data, the Alzheimer’s Prevention Initiative upped the crenezumab dose administered subcutaneously to the participants in its secondary prevention trial in Colombia to match the IV levels, Reiman said (see May 2012 conference story). Meanwhile, Genentech will continue its high-dose extension trial of crenezumab, and has met with the FDA to discuss whether to develop the antibody further, Salloway said.
Progress reports from other antibody trials focused on patient selection. Philip Scheltens of VU University Medical Center, Amsterdam, detailed baseline data for the ongoing Phase 3 SCarlet RoAD trial of Roche’s monoclonal antibody gantenerumab. Gantenerumab is also being tested in the DIAN-TU prevention trial (see Oct 2012 conference story; Sep 2013 news story). Scheltens’ talk reviewed how screening with the new Dubois criteria (see Part 1 of this series) works out in the reality of a multicenter trial. Roche’s trial enrolls about 800 people with prodromal Alzheimer’s disease. Partway into the screening, it delivers the Free and Cued Selective Reminding Test (FCSRT) to measure if the candidate has a hippocampal memory deficit, and those who meet the cutoff then undergo a lumbar puncture to determine if the deficit is due to underlying Alzheimer’s pathology.
Of 3,000 people screened, 2,290 fell out at some point. Eight hundred and twenty-one scored above the upper cutoff on the FCSRT. Of those who passed this step, 621 more subsequently failed the CSF test, meaning they had an episodic memory deficit but no CSF evidence of AD pathology. Of those 621, roughly two-thirds, or 413, had usable data in the system. Scheltens compared this group of 413 to the baseline trial population. He found that people who failed the CSF screen were younger than the trial population and had a milder cognitive phenotype. They scored better on global cognitive measures such as CDR, ADAS-Cog, and MMSE, as well as on tests of episodic memory and executive function, Scheltens said. Overall, the use of a CSF screen to enrich a prodromal population selected people with greater deficits, he concluded. On the other hand, if the researchers had not screened for amyloid, they would have enrolled those 621 people in the trial, meaning about 43 percent of the participants would have lacked evidence of amyloid pathology.
Similar results were reported on a poster from Jeff Sevigny, Biogen Idec, Cambridge, Massachusetts. For the Phase 1b trial of its therapeutic antibody aducanumab (aka BIIB037), the researchers first screened volunteers using cognitive measures such as the FCSRT, CDR, and MMSE, then measured PET amyloid burden in those who met the cognitive criteria. Out of the first 278 people scanned, about 40 percent had no brain amyloid, similar to Roche’s findings. Likewise, Lilly insiders said at CTAD that screening for the ongoing solanezumab trial in mild AD also fails a significant fraction of cognitively admissible candidates at the subsequent brain amyloid step.
By comparison, in the bapineuzumab and solanezumab trials and ADNI, the number of amyloid-negative participants ran around 15 to 20 percent. Current trials are enrolling an earlier, prodromal population, and thus may pick up more people with cognitive impairment not due to AD, Sevigny suggested in his poster. Supporting this, in Biogen’s screens ApoE4 carriers were more likely to accumulate amyloid, with 80 percent of them testing positive, compared to 43 percent of non-carriers. The findings underscore the importance of amyloid screening for these early trials, Sevigny concluded.
Perhaps the most unexpected passive immunotherapy news came after CTAD closed. Biogen announced on December 2 that it will push aducanumab straight from Phase 1b to Phase 3. The Phase 1 trial enrolled nearly 200 people with prodromal AD and was scheduled to conclude in 2017 (see Apr 2013 conference story). According to an interim analysis, one year of dosing lowered brain amyloid and improved cognition in participants, the company’s Doug Williams claimed at an investor conference. The data will be presented at a research conference next year.—Madolyn Bowman Rogers
Immunotherapy II: Active Approaches Down, New Passive Crops Up
While researchers pursuing passive immunotherapy approaches struggle to refine trial protocols (see Part 4 of this series), active immunotherapies presented setbacks at the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20 to 22 in Philadelphia. Speakers clarified baffling data from AFFiRiS’ Phase 2 trial of AD02, in which the placebo was said to beat the treatment. The treatment did not work and is not being pursued any more. Another active vaccine, Pfizer/Janssen Alzheimer Immunotherapy’s ACC-001, nudged biomarkers, yet the company has shelved development. There were no presentations at CTAD on CAD106, currently the most advanced active immunotherapy in AD. Passive approaches were the hotter topic here, with a new candidate— Eli Lilly’s LY3002813—making its debut.
AFFiRiS AG, Vienna, unfurled perhaps the most curious story of the meeting. At an earlier press conference, the company had reported that its placebo worked better than its drug, and announced plans to further develop the placebo, which it declined to identify (see Jun 2014 news story). In Philadelphia, the company’s Achim Schneeberger cleared up the mystery, revealing that the placebo was alum, an aluminum salt (typically potassium aluminum sulfate) frequently used as an adjuvant for vaccines. The researchers injected either 1 mg or 2 mg alum into the control cohorts, while the three treatment groups received either 1 or 2 mg of alum along with varying amounts of the active vaccine, AD02. The Phase 2 trial enrolled 332 people with early AD from six European countries.
Those who received 2 mg alum but no vaccine had the best outcomes. Huh? Yes, you read it correctly. This group reportedly declined less than the others, and did so consistently across several measures, including the ADAS-Cog, Activities of Daily Living (ADL), and Clinical Dementia Rating Sum of Boxes (CDR-SB). In fact, almost half of this control cohort remained cognitively stable over the 18 months of the trial, Schneeberger reported. Their hippocampi shrank less compared to the other groups’, correlating with higher ADAS-Cog scores. This cohort also stabilized on measures of neuropsychiatric symptoms and quality of life. Dropout rates were similar between the cohorts.
Several clinicians at CTAD privately suggested this may be a chance finding, but Schneeberger disagreed. “It is unlikely that the effect is by chance. We believe there is a disease-modifying effect,” he said. Some attendees questioned why, if alum protects, didn't 2 mg alum given in conjunction with AD02 slow decline? Schneeberger suggested that the AD02 peptide might interfere with the effect of alum by coating it.
Could the 2 mg alum group have declined as expected, while AD02 had a deleterious effect that made those taking it deteriorate faster than normal? Probably not, said Suzanne Hendrix, a statistician at Pentara Corporation, Salt Lake City, who works on contract with AFFiRiS. Hendrix presented analyses that compared the decline in this trial with decline in historical datasets including the Alzheimer’s Disease Neuroimaging Initiative (ADNI), the Alzheimer’s Disease Cooperative Study (ADCS), and a trial simulation tool developed by the Coalition Against Major Diseases (CAMD) (see Jul 2013 news story). All cohorts in the AFFiRiS trial declined cognitively and functionally at the expected rates—except the 2 mg alum group, Hendrix said. “The 2 mg alum group was the outlier,” she added. She subdivided the cohort by disease severity and found that only those with mild AD appeared to benefit from alum; moderate patients worsened at expected rates.
AFFiRiS’ next goal is to discern how alum might slow decline, Schneeberger said. Hendrix noted that the 2 mg dose was chosen to provoke an immune reaction at the injection site. Perhaps the adjuvant somehow slows Alzheimer’s disease by stimulating the immune system, she suggested. To explore this, AFFiRiS will look for changes in cytokine levels in patient samples. The company plans to launch a new confirmatory trial in Europe, Hendrix told Alzforum. A U.S. trial would face additional safety hurdles, because 2 mg alum exceeds the 1.25 mg limit set by the Food and Drug Administration. In Europe, alum is approved at higher doses and frequently used in immunotherapy for a variety of conditions. Adding to the puzzle, epidemiological studies have linked aluminum exposure to Alzheimer’s risk (see, e.g., Bondy, 2014). In fact, there has been longstanding debate about aluminum’s role in Alzheimer’s, and families have been discarding their aluminum pots and pans for years.
Meanwhile, AD02 itself provided no statistically significant benefit, despite earlier results (see Jun 2012 conference story). AD02 was designed to stimulate the immune system to make antibodies against Aβ. With active vaccines, the dose of injected peptide itself matters less than the titer of the antibodies the patient’s B cells generate in response. That antibody concentration, in effect, represents the exposure of the body to “active drug.” Schneeberger presented no antibody titer data at CTAD. He told Alzforum that most patients did mount an immune response, which varied with the dosage of AD02 and adjuvant. The company has terminated an extension study of AD02. Appearing puzzled by the data, the audience asked no questions after Schneeberger’s talk.
Another active vaccine has also reached the end of its development life. Janssen’s ACC-001, originally developed by Elan/Wyeth, is a peptide of seven amino acids from the N-terminus of Aβ (see Apr 2008 news story; Jun 2008 news story). At CTAD, Nzeera Ketter of Janssen presented results from the company’s Phase 2 trial in 125 volunteers with mild to moderate AD. Participants received six intramuscular injections of either placebo, 3 mg, or 10 mg ACC-001 with the adjuvant QS-21. More than 90 percent of participants in the active groups responded to the vaccine by making antibodies, with titers modestly higher at 10 mg than 3, Ketter noted. Safety was as expected, with 5.8 percent of the cohort developing the leaky brain blood vessels known as ARIA-E.
Also as expected, no significant difference in cognitive decline appeared in this small study. However, those on the drug did accumulate less brain amyloid as assessed by florbetapir PET imaging. This effect increased at the higher vaccine dose, but missed statistical significance. Plasma Aβ levels rose along with antibody titers, suggesting enhanced clearance from brain. In the treatment group, phosphorylated tau in the cerebrospinal fluid (CSF) dropped slightly, again hinting at a treatment benefit. The only statistically significant difference between the cohorts was more shrinkage in brain volume in the high-dose treatment group. Why this happens is unclear, but it mirrors what has been seen in other antibody trials, Ketter noted (see Jul 2004 conference story; Nov 2012 conference story). Though small, these biomarker effects demonstrate that the generated antibodies hit their target, Ketter said.
Janssen also tested the same doses of ACC-001 in an amyloid imaging and safety Phase 2 trial of 63 early AD patients. Presented on a poster at CTAD, those data were likewise negative overall, with subtle hints of biomarker movement. In response to an audience question, Ketter noted that Janssen has shelved this vaccine.
Even as some vaccines reach the end of the road, new hopefuls appear. Eli Lilly has an antibody against a modified form of Aβ in Phase 1. LY3002813, also known as N3pG, targets an N-truncated, pyroglutamate form of Aβ that aggregates readily and forms the core of many plaques. LY3002813 cleared deposits in mouse studies without producing microhemorrhages (see Dec 2012 news story). John Sims at Lilly described the trial. At four U.S. and two Japanese sites, the single-ascending-dose study will test intravenous injections of 0.1, 0.3, 1, 3, and 10 mg/kg, as well as subcutaneous injections of 3 mg/kg. Participants have a positive florbetapir scan, and range from MCI due to AD or mild to moderate AD as diagnosed with the AA-NIA revised criteria. In the first month, the most common adverse events were diarrhea, dizziness, and falls, but no changes in microhemorrhages (ARIA-H) and no ARIA-E, Sims reported. So far so good, but there are many more hurdles to jump.—Madolyn Bowman Rogers
New Target Has Legs: Tau PET, Mice, and Antibodies
Coming to the fore as both a disease marker and a therapeutic target, the protein tau drew its share of attention at the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20-22 in Philadelphia. Speakers presented fresh evidence that tau PET imaging reflects the stage of a person’s disease and cognition, suggesting that investigational tracers may allow researchers to track progression in clinical trials. Meanwhile, new animal data strengthened the notion that pathological tau comes in strains that spread through the brain in stereotypical patterns of distinct tauopathies. Would mopping up this extracellular tau halt the march of disease? The idea appears to hold water in animals, with a speaker describing a new monoclonal antibody that restored memory in tau mice. Researchers are trying to learn from past instances where treatment results in mouse models failed to translate to human trials, and one talk introduced a new model that expresses wild-type human tau along with mutant human APP. This mouse develops tangles, memory loss, and neuronal degeneration without amyloid plaques.
Mouse neurofibrillary tangles without tau mutation.
Tangles came up by introducing wild-type human tau into APP transgenic mice. The tangles were visualized by Gallyas silver staining, and were confirmed to contain both 3R and 4R tau like those in AD brain. [Image courtesy of Takami Tomiyama, Osaka City University.]
With more trials targeting people in prodromal stages of disease, researchers need better ways to monitor progression, said Reisa Sperling of Brigham and Women’s Hospital, Boston. Cerebrospinal fluid (CSF) biomarkers plateau around the time symptoms appear, and barely record ongoing change. “CSF tau is just not a dynamic marker,” agreed Paul Aisen of the University of California, San Diego. Moreover, CSF tau levels do not reflect brain anatomy, and it is not only the concentration of tau but where it spreads that matters, Sperling noted. Tau imaging allows researchers to track how tau creeps through the brain as disease worsens. Sperling presented preliminary data from scans using Eli Lilly’s tracer T807 that offered clues to how tau relates to amyloid deposition and memory loss.
In cognitively normal participants of the Harvard Aging Brain Study, tau began to accumulate around age 40 to 50. By age 65 the majority of people had some tau deposits in their medial temporal lobes, corresponding to a Braak stage of I or II. “As someone who is getting closer to this age, this is frightening,” Sperling said. Older age groups were likelier to have some tau in the neocortex as well, resembling Braak stage III or IV.
How does this relate to CSF biomarkers? In a cohort of about 30 people, high tau PET correlated with high CSF tau. It correlated even more strongly with low CSF Aβ42. What’s more, having low CSF Aβ42 predicted that there would be tau outside of the medial temporal lobe. “Amyloid may accelerate the cortical deposition of tau,” Sperling suggested.
While both amyloid and tau spread from brain region to brain region, Sperling pointed out that the two pathological proteins tend to affect the brain differently. Amyloid disrupts long-range brain networks such as the default mode network, whereas tau tends to gunk up local circuits in the medial temporal lobe. Coordinated activity between the default-mode network and the medial temporal lobe is critical to encode and then retrieve memories. “Perhaps the combination is what harms memory function,” she speculated.
Prior tau imaging studies have found that uptake of tau tracer correlates with early memory problems (see Nov 2013 conference news; Aug 2014 conference news), and more recent data bear this out. Sperling and Keith Johnson at Massachusetts General Hospital, Boston, by now have analyzed data from 93 people who had tau scans and memory tests within six months. Those with neocortical tau performed poorly, even if they had no brain amyloid, Sperling reported. “Tau is the more proximal mediator of cognitive decline,” she said. “Aβ tells you that you’re on the Alzheimer’s train, but tau tells you which stop you’re at.”
If a person has markers of both amyloid and tau, is it too late to intervene? In future prevention trials, Sperling plans to enroll people earlier on the amyloid accumulation curve, who do not yet have widespread tau. At later disease stages, patients might need a combination therapy that attacks both markers, she suggested (see Part 2 of this series).
Other data dovetailed with Sperling’s findings. Adam Schwarz of Eli Lilly in Indianapolis presented findings from a small study that compared T807 tau imaging patterns with Braak stages. The researchers defined eight regions of interest in temporal and occipital lobes based on sections typically used for staging. They scanned seven Alzheimer’s disease patients, five people with mild cognitive impairment, five age-matched controls, and four young controls. The profiles of where tracer bound in these diagnostic groups matched up well with published Braak patterns for these groups, allowing the researchers to categorize participants as stage I/II, III/IV, or V/VI, Schwarz reported. Most healthy controls had at least some tau in the entorhinal cortex, but this was counted as Braak stage zero until it reached a cutoff value. As expected, higher Braak stages correlated with a diagnosis of MCI or AD. Three of the 42 brain hemispheres scanned did not fit into a Braak stage. “We see an encouraging correspondence between typical patterns of Braak progression and T807 binding,” Schwarz said. Autopsy studies will be needed to confirm that the tracer binds pathological tau. Next, Schwarz plans to scan larger cohorts, and to map other tauopathies, such as corticobasal degeneration (CBD), with T807.
The patterns of tau progression suggest that it spreads along neural networks, an idea supported by many animal and cell culture studies (see Jun 2009 news; Mar 2013 conference news; Aug 2013 conference news). At CTAD, John Trojanowski of the University of Pennsylvania, Philadelphia, extended this literature, noting that the injection of small amounts of synthetic preformed tau fibrils into different brain regions of PS19 transgenic mice kicks off tau pathology that spreads to connected regions over time (see Iba et al., 2013). The tau aggregates formed this way resemble the neurofibrillary tangles seen in AD, which would not otherwise develop in these mice, Trojanowski added. Injections into the locus coeruleus, believed to be one of the earliest sites of tau accumulation, resulted in pathological tau spreading both retrogradely to afferent brain regions and anterogradely to efferent regions. In a sense, injection of tau fibrils maps a given area’s anatomical connectome, these studies suggest.
Are all tau aggregates equivalent? Emerging data suggests not. Trojanowski’s group injected tau extracts from postmortem AD and CBD brains into the hippocampus and cortex of young PS19 mice. The injections triggered distinct pathologies reminiscent of their human origin. Mice receiving CBD tau developed tau inclusions mostly in oligodendrocytes near the injection site, from where they spread gradually through white matter to the hippocampal fimbria, external capsule, and stratum radiatum, but not into cortex. CBD is marked by neurofibrillary tangles predominantly in oligodendrocytes, not neurons. Few neurons died in these animals. By contrast, mice injected with AD tau formed tau aggregates in hippocampal neurons, from where they spread to connected brain regions such as the entorhinal cortex and locus coeruleus, and eventually to thalamus and cortex. In these mice, up to 50 percent of neurons died in affected areas (Boluda et al., 2014). The data fit with other recent work hinting that tau can form distinct strains with different pathological characteristics (see Clavaguera et al., 2013; May 2014 news story).
If pathological tau travels between cells, that journey should open a space for antibodies to sweep up the fibrils. Takami Tomiyama of Osaka City University, Japan, told CTAD attendees that both active and passive tau immunotherapy work in mice. Passive approaches are less prone to causing neuroinflammation, he noted, but previous studies have mostly targeted early stages of the disease, before symptoms appear. Tomiyama wanted to develop an antibody to treat advanced disease. First, he studied the phosphorylation patterns of human pathological tau to select the best epitope to target. They picked the phosphorylated Ser413 residue and generated the Ta1505 antibody against it. In human postmortem brain, Ta1505 lit up AD brains but not controls, demonstrating specificity for pathological forms of the protein, Tomiyama said.
The researchers then immunized 14-month-old tau model mice with either Ta1505 or Ta4, which binds to phosphorylated Ser396, another pathological tau residue. Both antibodies recognize only pathological tau in AD brain, not normal tau in control brain, Tomiyama wrote to Alzforum. Mice injected with Ta1505 had less phosphorylated tau, neurofibrillary tangles, and neuron loss. Levels of a synaptic marker rose, and memory returned to wild-type performance, Tomiyama showed. Animals receiving Ta4 improved somewhat on memory tests and phosphorylated tau, but other markers did not change.
Ta1505 may be a promising prototype for humanized antibodies, Tomiyama said. A patent application has been filed, and the paper is in press in Annals of Clinical and Translational Neurology. For previous work along this line, see Aug 2007 news story; Nov 2010 conference story.
Existing tau mouse models have limitations, Tomohiro Umeda, also of Osaka City University, said at CTAD. Animals that overexpress human mutant APP get hyperphosphorylated tau but no neurofibrillary tangles. Double-transgenic mice that express both mutant human tau and APP do get tau tangles, but these animals do not truly model Alzheimer’s, because tau is not mutated in this disease. In human disease, Aβ oligomers are believed to initiate tau pathology by first activating GSK3β, which phosphorylates tau and causes it to aggregate. Why don’t tangles form in APP mice, then? Maybe the process needs human tau, Umeda suggested. He noted that human and mouse tau vary quite a bit at the N-terminal region, which may affect the protein’s ability to aggregate.
To test this idea, Umeda crossed APPOSK mice with tau264 animals. The APPOSK mice express human APP with the Osaka mutation, and have abundant Aβ oligomers but no plaques (see Apr 2010 news story). Tau264 mice carry a human wild-type tau transgene and express equal amounts of the 3R and 4R splice variants (see Umeda et al., 2013). Neither of these mouse models has tangles. The offspring, however, developed tau tangles in cortex and hippocampus at 18 months (see image above). They resembled human tangles and looked filamentous in the electron microscope, Umeda reported. The hybrids also developed more aggressive pathology than their APPOSK parents, with Aβ oligomers, phosphorylated tau, and synapse and neuron loss appearing at younger ages. The results demonstrate not only that tangle formation requires human tau, but also that Aβ oligomers suffice to trigger tangles, Umeda claimed (see Umeda et al., 2014).
Attendees reacted to the model with enthusiasm. Many researchers are trying to move away from overexpression mouse models in favor of more physiological expression levels. At a stem cell symposium held November 6 at Duke University in Durham, North Carolina, Frank LaFerla of the University of California, Irvine, described a mouse model he made that expresses wild-type human APP at native levels and accumulates Aβ plaques by 17 months of age. This animal might resemble sporadic AD more closely than current models do, he suggested.—Madolyn Bowman Rogers
Try This at Home: Cognitive Testing in the Age of Prevention Trials?
As Alzheimer’s treatment pushes into ever-earlier disease stages, researchers need better ways of finding people who are at risk for cognitive decline. They also need better ways of cheaply tracking cognition in those people over multi-year trials. At the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20 to 22 in Philadelphia, speakers discussed how to move cognitive testing into people’s homes to meet these needs. In the future, Internet tests and perhaps even games played by hundreds of thousands of gamers may flag those whose cognition is beginning to wane ever so subtly, potentially helping researchers recruit for prevention trials. Data from one study indicated that because this method picks out people who are already declining, it could boost the statistical power for detecting a preventive effect. A validation study determined that cognitive tests taken at home on an iPad gave the same results as those done under supervision at a clinic. That could allow for more frequent testing and better sensitivity to changes. Another researcher introduced a questionnaire that a patient or caregiver can complete at home in five minutes and that allows clinicians to generate a valid Clinical Dementia Rating (CDR) score. These new methods form part of a wave of innovation in Alzheimer’s trials (see Part 1 of this series).
For patients enrolled in trials, frequent clinic visits are burdensome. They are a major reason why participants drop out of trials, particularly as the patient declines and clinic visits overwhelm the caregiver. They also drive up costs related to clinic use and staff to supervise testing. Home-based testing could lift some stress off participants’ shoulders and make long, large prevention studies more economically feasible. But can tests taken at home reliably measure cognition? Distractions might arise during testing, or older adults might make mistakes without a test administrator to guide them. In practice, these tests actually perform quite well, said Michael Weiner of the University of California, San Francisco. UCSF runs the Brain Health Registry, which six months after starting up has enrolled some10,000 adults from the Bay Area and around the world. About half the registrants are older than 60, and many have a family history of Alzheimer’s, Weiner said. UCSF partnered with Cogstate to make some of its card-based cognitive tests available for use on the site.
At CTAD, Weiner spoke about analyzing data from 3,500 registrants who took those tests. He compared the mean and standard deviation scores for each age range to the values that Cogstate records in clinical trials. The numbers were so close, Weiner told Alzforum, that at first he accused his statistician of mixing up the data. In addition, people who took the tests at home were equally likely to finish all the tasks as those who tested in a clinic. Most participants reported that they find the home environment more relaxing, Weiner added. He found that registrants with subjective memory complaints or a family history of AD scored lower than age-matched peers. The data suggest that at-home testing can be as accurate as that done in the clinic, he noted.
So far so good. But is there a way to reach much larger numbers of older adults? The brain-training company Lumosity, based in San Francisco, California, has many millions of users who play its online games, providing a potentially rich source of cognitive data (see Jun 2014 news story). Weiner collaborated with the company to analyze data collected from people who play its Memory Match game, which is designed to test visual working memory. The game is not a validated cognitive test, Weiner cautioned, and the data are noisy, to say the least. They present many analytical challenges: People play intermittently, with long variable gaps between sessions, making it hard to compare scores from different players. Nonetheless, Weiner and colleagues believe that they can calculate a “learning rate” based on how much a person’s play improves on consecutive sessions closely spaced in time.
The researchers analyzed de-identified data from some 2,200 people aged 40 to 79 who had played at least 40 game sessions over about one year’s time. They found a large age effect, whereby older people posted lower initial game scores and learned at a slower rate. Nonetheless, the over-60 group did not decline noticeably over the course of the study period. This matches findings from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and a large body of cognitive aging research, where healthy controls do not decline cognitively over one or two years, Weiner noted.
Among these older adults, however, there were some whose learning rate dropped during the study. Based on their average rate of decline, Weiner estimated that a one-year study enrolling this group would need 202 people per arm to detect a 25 percent slowing of cognitive decline due to treatment. Thus, recruiting from this population could lower the sample size needed in a preventative trial, Weiner claimed. Such a method of recruitment could be cheap and provide access to a large pool of potential trial volunteers. “This is a proof of concept that using the Internet to assess cognition in longitudinal datasets can be useful in neuroscience research,” Weiner said.
Can cognitive tests taken at home on an iPad do away with frequent trips to the memory clinic? A validation study of face-name, card-sorting, and pattern-separation tests suggests they can. [Image courtesy of Dorene Rentz, Brigham and Women’s Hospital.]
But would tests taken at home be a practical, reliable way to collect cognitive data? An early study using technologies such as telephone interviews, mail-in questionnaires, and Internet-based kiosks found a high dropout rate in the technology groups, but also hinted that the home-based methods were time-efficient despite requiring more training in the beginning (see Sano et al., 2010). Since then, technology has become simpler, with tablets now quite user-friendly. To see if these devices could collect accurate data at home, Dorene Rentz of Massachusetts General Hospital, Boston, enrolled 39 cognitively healthy, highly educated participants with an average age of 72.
First, participants came to the clinic to take the paper-and-pencil version of the ADCS-PACC composite that is being used in A4 (see Jun 2014 news story and Part 3 of this series). At this visit, they also took version A of a computerized cognitive composite (C3) on an iPad. The C3 includes four tests from the Cogstate battery, as well as two novel episodic memory tests: a face-name associative memory task and a pattern-separation task. The C3 is also being explored in A4, but with staff supervision, not home-based. This study validated the home setting against clinic testing. At the end of the visit, participants took home the iPad, an instruction manual, and a phone number to call if they needed help. Over the next five days, they had to complete one alternate version of the C3 each day. At the end of the week, participants returned to the clinic to take version A of the C3 again.
How well did they do? Sixty-two percent completed all five tests correctly, and 95 percent completed four of five properly, Rentz reported. The biggest problem people had was taking the wrong version of the C3; most often retaking version A at home. Scores on different versions of the C3 agreed extremely well, indicating that these tests were equivalent, Rentz said. C3 scores from home and the clinic matched as well, with a correlation of 0.7, demonstrating that where someone took the test made no difference. The paper-and-pencil PACC results also correlated significantly with the C3. The statistical analysis was done with Samantha Burnham at CSIRO Computational Informatics in Floreat, Australia.
These results suggest that at-home tablet testing could be incorporated into trials, Rentz said. Next, she wants to see whether cognitively impaired people can complete this task, perhaps with help from a study partner. She will also try to set cutoff scores for normal versus impaired users, and study larger cohorts. Meanwhile, Cogstate is improving its program to help users select the proper test each time, Rentz said, and electronic fingerprinting as used in new mobile phones may help ascertain that it’s actually the proband, not the partner, who takes the test at home.
Rentz’s talk was enthusiastically received. Some scientists said they are setting up similar systems of home-based iPad tests where results are downloadable to the clinic; others emphasized that once the process is fully validated, being able to obtain much more frequent testing than is feasible with clinic visits will greatly improve the quality of cognition tracking. Steve Ferris of New York University Langone Medical Center noted that home-based iPad tests will be particularly useful in prevention trials of lifestyle interventions, which require large numbers of participants but far fewer clinic visits than trials of an invasive drug such as an antibody.
Can home-based tests also stage dementia severity? On a poster at CTAD, James Galvin of NYU Langone noted that gold-standard assessments for staging dementia, such as the CDR, require a trained clinician and take at least half an hour to complete. They work in trials, but not to screen large numbers of people. On the other hand, quick screening tests such as the AD8 stage disease poorly (see May 2007 Webinar). To find a middle ground, Galvin developed the Quick Dementia Rating System (QDRS). This questionnaire takes five minutes for either the patient or a caregiver to complete. It could be given over the Internet or taken with paper and pencil at home, Galvin said. The QDRS comprises 10 questions measuring domains such as memory, problem-solving, mood, and language, with scores ranging from zero for a cognitively normal person to 30 for severe impairment. The four cognitive and six behavioral domains match up closely with those measured by the CDR.
In initial testing in a cohort of 736 dementia caregivers, QDRS scores correlated highly with traditional rating scales, including measures of quality of life and caregiver burden. Galvin and colleagues then launched a validation study of the screen in 259 patient-caregiver pairs who get care at NYU. Caregivers filled out the QDRS at home before their clinic visit and dropped the form off as they came in. A blinded clinician then did a formal CDR, and Galvin compared how the two matched up. QDRS scores climbed with increasing CDR stage, with a correlation of r=0.73 between the two measures, the scientists reported at CTAD. In reliability testing, the QDRS had an ICC score of 0.93 with the CDR-sum of boxes. “By looking at the QDRS, I know what the person’s CDR-SB will be,” Galvin told Alzforum. The QDRS discriminated well between cognitively healthy people and those with mild cognitive impairment, Galvin reported. It also correlated with the Neuropsychiatric Inventory and Mini Mental State Exam.
The screen could be useful both to help recruit people for trials and for home-based assessment in longitudinal studies, Galvin suggested. He next plans to measure how people’s scores change over time. Like the CDR, the QDRS does not identify what disease underlies a person’s score—it could be AD, dementia with Lewy bodies, or something else.—Madolyn Bowman Rogers
Just for Her? Study of Women’s Biology Offers New Therapeutic Angle
It’s not news that women run a higher risk of Alzheimer’s disease than men. Nor is the thought that this has something to do with female sex hormones ebbing after menopause. Alas, hormone replacement has not worked, and it’s time for fresh ideas. Researchers from the University of Southern California, Los Angeles, explored new ways to bolster cognition via the hormone axis in older women in a symposium at the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20 to 22 in Philadelphia. They characterized a metabolic profile that may flag postmenopausal women at risk for cognitive decline. They also presented therapeutic approaches in clinical trials, one involving a food supplement made from plant estrogens and the other involving injections of the neurosteroid allopregnanolone to stimulate neurogenesis. If this sounds like a ladies-only approach, not so. These treatments may benefit men, as well, speakers said.
Neurosphere of neural stem cells. [Image courtesy of Robbie Brinton.]
Estrogen-based therapies have a checkered history. On the one hand, animal and cell culture studies have established that estrogen signaling in the brain, particularly through estrogen receptor β (ERβ), promotes remodeling of spines and synapses, boosts excitatory neurotransmission, and appears to dampen neuroinflammation (see Feb 2008 news story; Nov 2011 conference story; May 2011 news story). Epidemiological studies have linked hormone replacement therapy (HRT) with lower Alzheimer’s risk, and a small trial found that short-term hormone therapy maintained brain volume in postmenopausal women (see AlzRisk entry; Nov 2002 news story; Nov 2011 conference story). But subsequently, a finding of higher risk of dementia, heart disease, and some kinds of cancer with HRT in the Women’s Health Initiative threw cold water on the idea of estrogen-based therapies (see Jul 2002 news story).
Estrogen regulates how the brain uses energy, including glucose uptake and mitochondrial function. In animal studies, the loss of reproductive hormones changes brain metabolism, noted Roberta Brinton of USC. If this happens in older women, it might put them at risk of Alzheimer’s, since falling glucose use in the brain foreshadows the disease. Individual variation clearly plays a role, since all women go through menopause but only some head down the path toward dementia. How to find them? Working with Brinton, Jamaica Rettberg of USC is searching for early markers to identify those postmenopausal women most at risk for cognitive decline. To do this, she analyzed data from the five-year Early Versus Late Intervention Trial With Estradiol. ELITE comprises 643 women; almost half have recently gone through menopause, the remainder are 10 years or more past this milestone (see Hodis et al., 2014). Because peripheral metabolic biomarkers can serve as reporters of brain metabolism, Rettberg examined data on baseline metabolic measures such as blood glucose, insulin resistance, cholesterol, triglycerides, ketones, and blood pressure.
All the women had numbers in the normal range. Their data clustered into three distinct profiles, Rettberg said. Women with the “healthy metabolic” profile were at the lower end of normal, with an average blood pressure of 106/68, LDL cholesterol of 130, and fasting blood glucose of 80, whereas women judged to have a “poor metabolic” profile had numbers on the higher end of normal, such as a blood pressure of 121/76, LDL of 145, and blood glucose of 92. Women in this cluster also had much higher insulin resistance and triglycerides than their peers. The third group, the “high blood pressure” group, displayed a mixed profile, with high blood pressure and moderate insulin resistance and triglycerides. The study included about 40 percent minorities, and Hispanic women were more likely to have poor metabolism.
Rettberg then compared the cognitive performance at baseline of the three clusters. She saw a trend for the healthy group to score highest, the high blood pressure group in the middle, and the poor metabolic group lowest. This suggested to Rettberg an association between metabolism and cognition. Were the women with poor metabolic scores at greater risk for future dementia? To see how performance changed over time in the absence of hormones, Rettberg analyzed outcome measures for women on placebo. However, by the end of the study, all groups had improved significantly on measures of memory and global cognition, which Rettberg attributed to the women learning the tests. Tests of executive function showed no such practice effects; on these measures the high blood pressure group declined slightly.
Rettberg noted that all these metabolic measures are commonly used in routine medical care, hence the profiles could easily be adopted for screening. If a woman learns she may be at risk, can she avert dementia by changing her lifestyle? This is unknown. Over the five years of the study, most women remained in the same metabolic group, even those who improved on specific measures such as blood pressure. That said, the ELITE trial was not trying to change metabolism, Brinton noted. Also unknown is how hormone therapy changes women’s metabolic profiles. Rettberg will next analyze data from ELITE participants on estradiol to see whether hormone replacement make a difference. The researchers also want to see if the findings will translate to men.
Given concerns about standard HRT, what about safer alternatives? Some researchers have turned to estrogen-like compounds derived from plants. Known as phytoSERMs (selective estrogen receptor modulators), these compounds activate ERβ. PhytoSERMs are sold as dietary supplements, and some studies have reported cognitive benefits for postmenopausal women (for review, see Zhao and Brinton, 2007). At CTAD, Lon Schneider of USC detailed research into these compounds led by himself and Brinton. They found that a particular phytoSERM formulation, when given to female rodents whose ovaries had been removed, prevented menopause symptoms such as hot flashes, as well as waning brain metabolism and spatial memory performance (see Zhao et al., 2011; Yao et al., 2013). The phytoSERM formulation likewise improved spatial memory and prevented some amyloid plaques in female 3xTg AD mice when given before deposition began (see Zhao et al., 2013). The preparation consists of an equal mixture of three phytoSERMs from soy: daidzein, genistein, and S-equol.
Schneider described an ongoing Phase 1/2 trial of this preparation. He is trying to develop this formulation as a food supplement with a structure-function claim, not a drug. This allows for a nutraceutical regulatory path, which is cheaper. The study group consists of 71 women who are undergoing or recently underwent menopause, and who have hot flashes and concerns about their memory. They take one tablet per day, either placebo or 50 mg or 100 mg of phytoSERM. The 12-week study includes a crossover at four weeks, i.e., some participants switch from placebo to drug or vice versa. This design boosts the power of small studies to detect a treatment effect, because each participant provides her own drug-placebo comparison. Outcomes include memory tests, a neuropsychological battery, measures of menopause symptoms such as hot flashes, and markers of mitochondrial function. In answer to an audience question, Schneider noted that previous studies have shown that phytoSERMs in general enter the brain. In this trial he will measure only plasma concentrations, not CSF, and so will not have direct proof that this particular formulation crosses the blood-brain barrier.
Data collection will wrap up this month, Schneider said. If he sees a consistent cognitive benefit, i.e., a benefit across multiple measures, Schneider said he would consider applying for an Investigational New Drug (IND) authorization for future trials, which could include men as well.
Beyond phytoSERMs, the CTAD audience heard an update on allopregnanolone. “This is a first-in-class regenerative therapeutic to treat early Alzheimer’s,” Brinton told the audience. In mouse models, this neurosteroid metabolite of progesterone triggers the birth of new neurons, curbs amyloid deposition, and preserves cognition (see Oct 2005 conference story; Mar 2010 news story).
Brinton and Schneider are leading a Phase 1 multiple-ascending-dose trial of the steroid in 16 men and 16 women with MCI due to AD or mild AD (see Aug 2013 conference story). The trial is enrolling participants, and Brinton expects to begin dosing in January 2015. She also told Alzforum that she had just finished analyzing toxicology data from a six-month allopregnanolone study in rats, and the data indicate good safety for chronic treatment.
During the trial, the researchers will scan participants with MRI to detect any microhemorrhages due to treatment. This is a requirement from the Food and Drug Administration, but Brinton noted that the MRI data will also allow her to monitor for possible brain regeneration by measuring hippocampal volume, brain connectivity, and white-matter tracts.
Will these approaches leave men out in the cold? Far from it, researchers say. Men’s brains also express ERβ, hence they might benefit from treatments that tickle this pathway. Allopregnanolone is already being tested in both men and women, and the metabolic profiling to identify populations at risk for AD that was first developed in women could be used to also identify men at risk. As the saying goes, what is good for the goose may yet be good for the gander.—Madolyn Bowman Rogers
New Ideas for Alzheimer’s Treatment: What’s on Offer in 2015?
Researchers agree that a disease as complex as Alzheimer’s may require combination therapies, but so far, few trials have taken on this challenge. That may soon change. At the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference in Philadelphia November 20 to 22, an impassioned case for developing multiple investigational compounds together came from none other than Rusty Katz, who last year retired from the Food and Drug Administration (see Part 2 of this series). Also at CTAD, Jean-Marc Orgogozo of University Hospital Pellegrin, Bordeaux, France, presented one example of a combination treatment, though not of investigational drugs. He reported that a combination of two repurposed drugs, neither of which has any efficacy for AD on its own, improved cognition in AD patients over a 12-week trial. “I’m delighted to hear of the evolution in the FDA’s thinking,” Orgogozo said.
That said, his trial is the exception. For the most part, CTAD featured single therapies directed at a variety of targets. Taken together with an update on European trials of non-pharmacological interventions, the meeting showcased a field trying to implement calls to broaden therapeutic approaches beyond amyloid and tau. Below, a summary.
In collaboration with the pharmaceutical company Pharnext in Issy-les-Moulineaux, France, Orgogozo led the PLEODIAL I study to test PXT00864. This compound mixes low doses of acamprosate calcium, used to treat alcohol dependency, and the muscle relaxer baclofen, approved for spasticity. Both drugs tweak neurotransmission. Acamprosate is an analog of the neurotransmitter GABA, while baclofen activates the GABA-B receptor. Together, the drugs appear to suppress excitatory and boost inhibitory signaling, Orgogozo said. This would appear to be an opposite effect to idalopirdine, which is currently in Phase 3 (see Part 10 of this series). Orgogozo believes the PXT00864 combination may help restore balance to AD brains, which are prone to hyperexcitability (see Sep 2007 news story). In two different AD mouse models, the combination restored working memory and object recognition to wild-type levels, and kept neurons alive, Orgogozo reported. When tested separately, neither drug had any effect.
The Phase 2a PLEODIAL trial enrolled 30 people with mild AD. Each participant took one of two doses of PXT00864 for four weeks, switched to placebo for four more weeks, then returned to the drug for the remainder of the trial. This design increases the trial power because each participant acts as his or her own control. While on the drug, participants improved on the ADAS-Cog11, whereas they declined while on placebo, Orgogozo said. The researchers saw a trend toward improvement on tests of executive function as well, but the scores were more variable and did not reach significance, Orgogozo said. Patients tolerated the drug well, mainly reporting some dizziness and fatigue at the higher dose. The researchers are running an extension trial of PXT00864, and plan next to test three different doses of the drug in a six-month Phase 2 study. A poster presented by Pharnext scientists suggested that in 20 healthy, young, male volunteers, PXT00864 enhanced recovery from a temporary memory impairment induced by scopolamine, an antagonist of cholinergic transmission that is often used in memory research.
Resveratrol, spinning in vain to get into the brain?
While PXT00864 appears to act via neurotransmission, the polyphenol resveratrolmay act through multiple different mechanisms. Resveratrol occurs naturally in foods such as red grapes and dark chocolate, and is selling briskly as a supplement. It has been variously reported to activate deacetylases known as sirtuins, which are believed to extend life; have antioxidant properties; and prevent amyloid deposition (see Jun 2010 news story; Sep 2013 news story). Several studies are testing this compound in people with cognitive impairment or dementia, and researchers recently reported that six months of resveratrol supplements improved memory and brain metabolism in a small trial of overweight, cognitively healthy older people (see Jun 2014 news story).
At CTAD, Scott Turner of Georgetown University, Washington, D.C., reported on the 12-month Phase 2 Alzheimer’s Disease Cooperative Study (ADCS) of resveratrol. In this trial, 120 AD patients at 20 ADCS sites started on 500 mg per day of synthetic resveratrol, gradually increasing to 2,000 mg per day. The compound reached maximum microgram concentrations in the blood within two hours and persisted for many hours, Turner reported. However, less than 1 percent of resveratrol and its metabolites entered the brain, with cerebrospinal fluid (CSF) concentrations topping out at the nanomolar level. Participants taking the supplement lost about one kilogram of weight during the study, but otherwise tolerated the compound well.
As with many recent trials, the researchers saw more movement on biomarkers than cognition or function. Only one clinical outcome reached significance, with the treatment group better able to perform activities of daily living than the placebo group. Turner noted that the trial was not powered to detect clinical improvement, and he did not expect to see change there. On the other hand, CSF Aβ40 stabilized in the treated group, while it dropped by about 14 percent in controls. In plasma, Aβ40 dropped by 6 percent in participants on resveratrol, but plummeted by 20 percent in controls, Turner reported. A similar trend occurred with Aβ42 Turner said. To him, the data suggest that resveratrol engaged its target.
The steep decline in Aβ40 in the control group over the course of one year was soon questioned by the audience. In AD patients, CSF Aβ levels normally bottom out around the time symptoms begin (see Jan 2012 news story; Jul 2012 news story), though Turner noted that one study reported continued decline as dementia worsened (Nitsch et al., 1995). Few studies have charted longitudinal Aβ changes in plasma, but what evidence does exist suggests that there are no consistent changes in these markers (see Donohue et al., 2014).
In the ADCS trial, people on resveratrol lost more brain volume than those on placebo, an effect that has been seen in several immunotherapy trials. Turner noted that resveratrol can dampen inflammation, which might explain the shrinkage.
X-ray crystal structure of VX-745 bound to p38 MAPK alpha.
[Image courtesy of Vertex Pharmaceuticals.]
A repurposed compound with anti-inflammatory properties appears headed for clinical trials. John Alam, who started the virtual biotech company EIP Pharma in Cambridge, Massachusetts, introduced the CTAD audience to VX-745, a small molecule that inhibits the mitogen-activated protein kinase, aka p38 MAPK alpha (see Duffy et al., 2011). This enzyme stimulates release of the inflammatory cytokines TNFα and IL-1β from microglia (see Yasuda et al., 2011). In animal models, VX-745 appears to shift the balance from pro-inflammatory M1 to phagocytic M2 microglia, in the process improving mitochondrial function, synaptic transmission, and memory, Alam said. In 2-year-old Tg2576mice treated with 3 mg/kg for two weeks, amyloid plaque load dropped by half, perhaps through enhanced microglial clearance, Alam suggested. This research was done at the CRO Charles River Laboratories in Kuopio, Finland.
Because these mice develop little neuroinflammation, the researchers tested the drug on aged rats, which develop more. Three weeks of treatment at the highest dose tested, 4.5 mg/kg, lowered IL-1β levels while the postsynaptic marker PSD95 trended higher. The drug boosted cognition, with the rats that received 1.5 mg/kg faring best and performing like youngsters in the Morris water maze, Alam said (see Alam, 2014).
VX-745 is an old drug that was discovered at Vertex Pharmaceuticals, where Alam worked at the time (see Haddad, 2001). It was being tested for rheumatoid arthritis, but Vertex discontinued this work about a decade ago, reportedly in part due to adverse effects in the central nervous system (see news release).
Patients in those studies took 250 mg twice/day, which according to Alam equated to 10 mg/kg in rats. Alam has filed for a patent on using VX-745 for Alzheimer’s disease. He will first conduct a Phase 2a trial of VX-745 in patients with prodromal AD at VU Medical Center in Amsterdam, with Philip Scheltens of VU University Medical Center, Amsterdam, as principal investigator; this trial will start screening patients in early 2015. The second trial, to be done in the United States, is still being planned. Participants would take either 125 or 40 mg twice daily. The former is equivalent to the dose that suppressed inflammation in rats, the latter to the dose that best sharpened their cognition. Alam noted that VX-745 preferentially enters the brain, reaching concentrations almost twice those in plasma.
Finding effective therapies is not all about new drugs. Sandrine Andrieu, an epidemiologist at INSERM, Toulouse University, France, told CTAD attendees that several small studies of diet, exercise, and cognitive stimulation have shown promise in bolstering cognition or delaying AD, but need validation in large, biomarker-supported studies. The European Dementia Prevention Initiative (EDPI) coordinates three large prevention trials (see Jun 2012 conference story; Sep 2012 news story), each of which enroll more than 1,000 older adults at risk of dementia and provide intensive health and cardiovascular treatments over the course of several years. One of these, the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), recently reported better cognition in seniors who followed its plan (see Jul 2014 conference story).
The findings have stoked interest in this area. EDPI has begun a pilot study called Healthy Aging Through Internet Counseling in the Elderly (HATICE) to see if information given over the Internet can help healthy older people ward off dementia. Another prevention study, Do-Health, just finished recruiting 2,000 adults 70 and older from seven European cities. The trial will test whether Vitamin D, omega-3 fatty acids, and exercise can help maintain cognitive performance.
These trials have limitations, Andrieu noted. Volunteers tend to be highly educated and may not represent the general population. Because they score quite well on cognitive tests at baseline, there may not be much room for improvement. Screening volunteers for brain amyloid deposition could enrich for those at higher risk, she suggested. Prevention trials might also need to run longer than the current ones do to demonstrate an effect. For example, participants in the Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE) study improved over the control group only after five years or more (see Rebok et al., 2014). Long trials are expensive and suffer high attrition, however. In addition, researchers should keep in mind that different interventions may work better for different groups of people, Andrieu said.
Despite these challenges, Andrieu stressed the importance of developing these approaches along with pharmaceutical options. “Even a small individual benefit can have a huge effect at the population level,” she said.— Madolyn Bowman Rogers
Duffy JP, Harrington EM, Salituro FG, Cochran JE, Green J, Gao H, Bemis GW, Evindar G, Galullo VP, Ford PJ, Germann UA, Wilson KP, Bellon SF, Chen G, Taslimi P, Jones P, Huang C, Pazhanisamy S, Wang YM, Murcko MA, Su MS.
The Discovery of VX-745: A Novel and Selective p38α Kinase Inhibitor.
ACS Med Chem Lett. 2011 Oct 13;2(10):758-63. Epub 2011 Jul 28
PubMed.
Shineman DW, Alam J, Anderson M, Black SE, Carman AJ, Cummings JL, Dacks PA, Dudley JT, Frail DE, Green A, Lane RF, Lappin D, Simuni T, Stefanacci RG, Sherer T, Fillit HM.
Overcoming obstacles to repurposing for neurodegenerative disease.
Ann Clin Transl Neurol. 2014 Jul;1(7):512-8. Epub 2014 Jun 24
PubMed.
New Treatments for Alzheimer’s Behavioral Symptoms on Horizon
In the apparent rubble of Alzheimer’s trials there is one area of therapy development that appears to offer modest hope. Several drugs for the treatment of neuropsychiatric symptoms have met endpoints in Phase 2 and 3 trials. Thus far, these compounds look safer than the atypical antipsychotics previously used to treat the mood and behavior problems that torment both patients and their caregivers. The new crop of neuropsychiatric drugs took center stage at the 7th Clinical Trials on Alzheimer’s Disease conference November 20 to 22 in Philadelphia, with two of the four symposia devoted, somewhat redundantly, to the topic. Speakers hailed the progress to date as potentially leading to a new era when psychiatric symptoms will be better managed, improving quality of life for patients and caregivers. The data generated a sense of momentum at a conference that otherwise delivered few major trial results, as most other large AD trials are a ways away from reading out. Jacques Touchon of University Hospital of Montpellier, France, who co-organizes CTAD, singled out the neuropsychiatric talks as a highlight of the meeting.
“Over the next three years, trials may establish a new treatment paradigm for agitation in AD,” predicted Susan Abushakra of Transition Therapeutics, San Mateo, California. “To me, this conference had the flavor of a new approach to treating this aspect of dementia,” said Michael Rafii of the University of California, San Diego, who is evaluating one such drug in adults with Down’s syndrome.
The need for these medications is great. Most Alzheimer’s patients have at least one neuropsychiatric symptom sometime during their illness, said Constantine Lyketsos of Johns Hopkins University, Baltimore. Besides agitation and aggression, these include depression, apathy, anxiety, emotional lability, and delusions. For caregivers, these are more distressing and difficult to deal with than the disease’s signature memory loss. They are the leading reason caregivers place loved ones in nursing homes (for a caregiver perspective, see Aug 2014 book review). For example, it is a fairly typical scenario in Alzheimer’s care that a wife will devote years of her life to caring for an increasingly incapacitated spouse, but when he tries to strangle her in a confused rage she commits him to institutional care. Behavioral problems add an estimated $10,000 per person per year to care costs. Moreover, patients with psychiatric symptoms progress faster to severe dementia and die earlier than people without these symptoms, Lyketsos said (see Rabins et al., 2012).
There are no good treatments. Atypical antipsychotics are used, but they sedate patients, can cause movement problems, and carry a black-box warning from the Food and Drug Administration because they increase the risk of death (see Oct 2005 news story; Jan 2009 news story).
Researchers are testing new and repurposed drugs with different mechanisms of action. Unlike atypical antipsychotics, which block dopamine receptors, the new compounds act on serotonin, acetylcholine, and norepinephrine. In Alzheimer’s, these neurotransmitter systems malfunction, Abushakra said. Acetylcholine transmission drops the most, by 50 to 85 percent, but serotonin, dopamine, and norepinephrine signaling are also suppressed. Paul Rosenberg of Johns Hopkins noted that neurotransmission goes haywire in patterns that correspond to particular psychiatric symptoms. For example, in agitated patients, hippocampal serotonin falls while dopamine turnover in the cerebellum rises (see Vermeiren et al., 2014; Vermeiren et al., 2014). The hope is that the new drugs may help to restore balance to brain signaling, speakers said.
Gentler Ways to Tame Agitation
Several current drug programs target agitation. Patients with this symptom become hyperactive and pace ceaselessly. Some lash out at caregivers, punching or choking them. Agitated patients may yell or curse in public places. They resist dressing or bathing. Some refuse to swallow food and medications in the belief that caregivers are trying to poison them. These symptoms not only put caregivers through the wringer, they also challenge trial personnel, who have to medicate and evaluate participants.
That said, it’s not just the patients. In the past, Lyketsos said, these studies were unable to demonstrate therapeutic effects because the outcome measures were inadequate, the placebo effect strong, and there was no good clinical definition of agitation (see Soto et al., 2014). To improve this, the International Psychogeriatric Association recently approved new criteria for defining this condition (see Cummings et al., 2014). Trials now are applying more detailed outcome measures and incorporating design features such as run-in periods and crossover from placebo to drug to minimize placebo effects. Several of these new studies are reporting successful results.
CTAD attendees voiced excitement about Avanir Pharmaceuticals’ AVP-923, a combination of the cough syrup ingredient dextromethorphan and quinidine to improve dextrometrophan’s pharmacology. Trade-named Nuedexta, this is the only medication approved to treat pseudobulbar affect. PBA is a socially embarrassing condition where people burst into uncontrolled crying and laughing; it occurs in several neurodegenerative diseases. It is rare in AD, but the drug may hold other benefits for dementia patients. At CTAD, Avanir’s Joao Siffert said that patients with amyotrophic lateral sclerosis and multiple sclerosis who took AVP-923 for PBA became noticeably less agitated. This led the company to test their drug in agitated dementia patients, where it met its endpoints in Phase 2 and is now headed for Phase 3 (see Oct 2014 conference story). Like other drugs under investigation for agitation, AVP-923 affects multiple neurotransmitter systems, inhibiting particularly reuptake of norepinephrine to enhance its signaling.
Other drugs are also posting encouraging results. Earlier this year the antidepressant citalopram, a selective serotonin reuptake inhibitor, was reported to dampen agitation (see Feb 2014 news story). In the Phase 3 CitAD study of 186 people with Alzheimer’s and agitation, those who took the drug for nine weeks improved. However, a worrisome safety issue emerged as well. The maximum dose of 30 mg/day slightly slowed down the heart by lengthening the “QT interval,” a portion of the heart’s normal rhythm. This change is believed to heighten the risk of tachycardia and sudden cardiac death. The FDA recommends that older adults take no more than 20 mg/day citalopram for any indication. At CTAD, Daniel Weintraub of the University of Pennsylvania, Philadelphia, said that in CitAD, the QT interval lengthened by about 18 milliseconds in patients on 30 mg/day citalopram. This is significant because an increase of 20 milliseconds or more is associated with risk of arrhythmia. Nonetheless, other researchers were optimistic about the drug, noting that adjusting the dose might alleviate this problem. Rosenberg said that to his mind, the QT prolongation is a question mark, not a stop signal. “I see the glass two-thirds full,” he said.
Anton Porsteinsson of the University of Rochester, New York, was an investigator of the CitAD trial. He told Alzforum that he is planning a new study to clarify whether lower doses of citalopram will control agitation without cardiac effects. He also wants to evaluate longer treatment. Weintraub reported that many patients, particularly younger people, improved only late into the trial. Conveniently for the manufacturer, this would imply that more people might benefit if they stayed on drug for longer periods of time.
Meanwhile, researchers are squeezing every last drop of data out of the Phase 3 study, but they were not forthcoming with it at CTAD. Weintraub said that six more CitAD papers will appear soon, on how genetic and other factors predicted patient response, and how blood levels of drug related to behavioral outcomes.
Elan Pharmaceuticals’ ELND005, also known as scyllo-inositol, appears to act via a different pathway. Scyllo-inositol has been reported to prevent Aβ aggregation and was initially tested for cognitive benefits in AD patients, but missed primary endpoints (see Aug 2010 news story; Aug 2011 conference story). In this trial, too, patients on the study drug seemed less agitated (see Jun 2012 conference story). Why might this be? Scyllo-inositol is an isomer of myoinositol, an intracellular messenger that kicks off the inositol trisphosphate (IP3) signaling pathway. In cell culture, scyllo-inositol lowers neuronal uptake of myoinositol, helping dial down the IP3 pathway, said Abushakra, who led development of the compound while at Elan. AD patients with psychiatric symptoms have high levels of myoinositol; other mood-stabilizing drugs, such as lithium, also lower myoinositol, she added.
Elan soon began a 12-week Phase 2 study of scyllo-inositol for agitation. The dose for the trial was selected to cut brain myoinositol levels by half. Participants will take 1,000 mg daily for four weeks in an initial “loading phase” of the trial, then 250 mg daily for eight weeks for maintenance. The trial will enroll 400 AD patients; 280 have signed up so far, Abushakra said. The main outcome measure will be change on an expanded version of the Neuropsychiatric Inventory agitation subscale. Whereas the older NPI-A assessed agitation and aggression with a combined eight items, the modified NPI-C subscale assesses agitation with 13 items and aggression with another eight questions. Thus, the NPI-C includes more specific behaviors and may be more sensitive, Abushakra said.
In addition to these compounds, the antihypertensive drug prazosin has entered a trial for agitation in AD after promising results in a small pilot study (see Jan 2013 news story). A poster at CTAD showed results from the pilot, noting that patients who did not respond to the drug had a drop in blood pressure when standing up, whereas those who responded did not. This difference might serve as a marker of therapeutic response, the authors suggested.
Other candidates in this crowded field include the dopamine receptor agonist brexpiprazole, which is in Phase 3 testing for agitation in Alzheimer’s. Researchers are also testing the oral cannabinoid Namisol in a Phase 2 trial to see if it can ameliorate behavioral problems in Alzheimer’s patients.
Can Neuropsychiatric Drugs Boost Memory While Soothing Anxiety?
Some companies hope that their neurotransmitter-tweaking drug can calm a person’s fears while also sharpening his or her thinking. Lundbeck’s idalopirdine, an antagonist of the serotonin 5-HT6 receptor that was originally developed by Eli Lilly, potentiates the effects of acetylcholinesterase inhibitors and facilitates central neurotransmission, Alireza Atri of Massachusetts General Hospital, Boston, said at CTAD. The compound appears to stimulate excitatory glutamatergic transmission and suppress inhibitory GABAergic signaling, suggesting that it shifts the balance in the brain toward excitatory activity, Atri said.
In Phase 2 trials of Alzheimer’s disease, idalopirdine added to the acetylcholinesterase inhibitor donepezil improved cognitive scores more than donepezil alone (see Jun 2012 news story; Oct 2014 news story). The combination lowered anxiety, and other psychiatric symptoms trended lower, Atri said. This drug was originally developed for schizophrenia but switched over to Alzheimer’s.
Three Phase 3 studies in mild to moderate AD are now ongoing, flourishing names such as Starbeam, Starshine, and Starbright. (Increasingly in pharma, Phase 3 studies get their own names, making individual trials sound like marketed products.) There is a Phase 3 extension study, as well. These studies will look for effects on both cognition and anxiety, Atri said. Pfizer has a similar 5-HT6 antagonist, PF-05212377, currently in a Phase 2 trial that will look for both cognitive and neuropsychiatric change.
What about other serotonin receptors? A poster at CTAD introduced Intra-Cellular Therapies Inc.’s ITI-007, an antagonist of the serotonin 5-HT2A receptor. At increasing doses, this antipsychotic compound also affects dopamine receptors, enhances glutamatergic transmission, and inhibits serotonin reuptake, the researchers claim. “The data are fairly clear that it is difficult to see efficacy in CNS diseases with molecules that target only one receptor,” the company’s Robert Davis told Alzforum.
The compound improved symptoms in schizophrenics in a Phase 2 study, and it is now in tests for AD. In a small Phase 1/2 safety study, 30 cognitively normal adults and eight Alzheimer’s patients tolerated seven days of dosing with up to 30 mg ITI-007. The trial was not powered to detect cognitive change, but the researchers reported improved word recall in both healthy elderly and dementia patients on the drug. Agitation could not be assessed because the AD patients did not have this symptom. The researchers are preparing for a larger Phase 2 study to start next year, which will measure behavior as a primary outcome and cognition as a secondary.
While the main focus at CTAD revolved around agitation in Alzheimer’s, drugs for other indications and patient groups are inching forward as well, said Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas. For example, ACADIA Pharmaceuticals’ pimavanserin, which selectively dials down the activity of the serotonin 5-HT2A receptor, met its endpoints in Phase 3 trials for psychosis in Parkinson’s patients (see Nov 2013 news story) and is being submitted for FDA approval. After gaining Breakthrough Therapy status (see MarketWatch), pimavanserin is now in a Phase 2 study for psychosis in AD, as well as a Phase 2 trial in schizophrenia.
The antidepressants paroxetine (Paxil) and venlafaxine (Effexor) each improved depression in Parkinson’s patients in Phase 3 (see Richard et al., 2012), and the stimulant ritalin relieved apathy in Alzheimer’s patients in Phase 2 (see Rosenberg et al., 2013).
Researchers are also turning their attention to people with Down’s syndrome, who are often overlooked in research. Carrying an extra copy of the amyloid precursor protein on their triplicated chromosome 21, people with Down’s develop Alzheimer’s pathology by middle age. This population also has behavioral symptoms of dementia, which tend to be the reason their distressed caregivers bring their loved ones to the clinic, said Rafii. At CTAD, Rafii reported results of a four-week Phase 2 trial of scyllo-inositol in 24 participants with Down’s syndrome. The trial had two active arms, at 250 or 500 mg daily. In the low-dose arm, plasma concentrations of the drug remained well below the efficacious dose, but in the high-dose arm, drug concentrations started overlapping with the efficacious range. Participants on this dose were less severely agitated, Rafii reported, but this initial trial was not powered to evaluate efficacy. The drug was well-tolerated, clearing the way for future studies to test higher doses and longer time frames, he added.—Madolyn Bowman Rogers
Large Studies Agree: Brain Amyloid Accelerates Cognitive Decline
With the advent of PET amyloid imaging, researchers discovered that as many as a third of cognitively healthy older adults go about their business with brains loaded with amyloid. What does this mean for their cognitive future? Are all these people destined to develop Alzheimer’s disease? Initial studies suggested that brain amyloid greatly ramps up the risk for cognitive decline and a clinical diagnosis of dementia, but it was clear that settling the question would take more years of observational study in larger numbers of people. At the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference in Philadelphia November 20 to 22, speakers expanded the body of evidence, presenting data from large prospective studies.
All the researchers reported that in their samples of otherwise healthy older adults, people with high brain amyloid levels declined cognitively over periods of two to three years, while those without brain amyloid remain stable. To be sure, the rate of cognitive decline varied from person to person, and some people with brain amyloid did stay sharp over these short time frames. It remains unclear whether these people are simply early in the trajectory of disease, or protective factors operate in their brain to help preserve cognition. Speakers discussed some factors that accelerate or slow disease progression, including ApoE genotype and hypertension. Tantalizingly for trial designers, deeper analysis of the data pouring in from prospective studies of preclinical and prodromal stages suggest that people’s decline on biomarkers and clinical measures during this phase happens in a linear fashion, simplifying the necessary statistics for both disease modeling and trial design.
Regardless of ApoE genotype, older AIBL participants without brain amyloid stayed cognitively stable over 4.5 years. Among those with brain amyloid, ApoE4 carriers declined much faster. [Image courtesy of Paul Maruff.]
Several studies have found that brain amyloid associates with lower baseline cognitive scores and faster decline (see Snitz et al., 2013; Mar 2014 news story), particularly when it occurs along with a marker of neurodegeneration (see Sep 2014 news story). However, these studies tended to be small, enrolling 200 people or fewer.
To see if the link would hold up in a larger sample, Ming Lu of Avid Radiopharmaceuticals, a subsidiary of Eli Lilly, analyzed data from 1,017 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) who had undergone a florbetapir PET scan. Lilly manufactures florbetapir. Almost half of the participants had been diagnosed with mild cognitive impairment (MCI) at baseline, and the remainder were either cognitively normal or diagnosed with Alzheimer’s disease. At CTAD, Lu reported that over three years, participants whose brains were free of amyloid stayed stable on cognitive tests, including the ADAS-Cog11, MMSE, and CDR-SB. In contrast, people with positive amyloid scans declined, with MMSE scores diverging significantly from amyloid-negatives at six months, and ADAS-Cog11 scores at 24 months. Moreover, for people with MCI, those with brain amyloid had almost five times the odds of progressing to AD over the course of the study than those without. However, Lu noted that brain amyloid explained only some of the individual variation in decline.
What determines how fast a given person declines? Lilly’s Paula Trzepacz combed through 24-month follow-up data from about 1,200 ADNI participants to hunt for clues. She used statistical analyses to look for subpopulations with different rates of decline, and identified four separate groups. The 470 people without brain amyloid fell into two categories of unequal numbers. About 90 percent remained cognitively stable, and they were equal parts cognitively normal or diagnosed with MCI. The remaining 10 percent of the amyloid-negatives surprised Trzepacz. Their cognitive scores dropped sharply over the study period. Most had been clinically diagnosed with MCI or AD; however, given the absence of amyloid it is likely that this was incorrect, and they suffered from a non-amyloid form of neurodegeneration, Trzepacz suggested. Other studies have identified people with “suspected non-amyloid pathology” (SNAP) as well (see Aug 2013 conference story; Nov 2013 news story; Nov 2014 news story). This potential SNAP group also had somewhat different symptoms from typical AD, having more difficulty with daily activities than memory, Trzepacz noted.
Trzepacz showed that the 722 people with brain high amyloid fell into two groups. One-quarter of them declined significantly during the study; most of these people had been diagnosed clinically with AD, some with MCI. The remaining three-quarters declined only slightly on the cognitive measures. They included mostly people who had been clinically diagnosed with MCI, and close inspection showed that they had higher cognitive scores at baseline. This cohort may be earlier in the trajectory of Alzheimer’s disease, Trzepacz suggested. The factor that best predicted rapid cognitive decline among amyloid-positive people was a low cognitive score at baseline, pointing perhaps to cognitive reserve as protection against the long-term cognitive effects of brain amyloid. Overall, the presence of amyloid does affect the trajectory of cognitive decline, and this occurs independently of the person’s diagnosis, Trzepacz concluded.
Paul Maruff of Cogstate in Melbourne, Australia, showed data from the Australian Imaging, Biomarker, and Lifestyle Flagship Study of Ageing (AIBL) that supported the hypotheses that factors in addition to amyloid level influence the rate of cognitive decline in early AD. AIBL is one of several prospective imaging and cognition studies that are generating empirical data to test and flesh out the hypothetical Alzheimer’s biomarker staging scheme. AIBL data show that once a person crosses the threshold of amyloid positivity, additional amyloid accumulates at a linear rate for about the next 20 years until Alzheimer’s symptoms appear. Importantly, this finding allows researchers to use linear models to predict progression in prodromal stages—the time period on which preventions trials increasingly are going to focus. “Think of the otherwise sigmoid Jack staging curves without the little bit at the beginning and the little bit at the end,” Maruff quipped.
Maruff analyzed AIBL data from some 200 healthy controls and 50 people with MCI who were being followed over three years. People without brain amyloid remained cognitively stable during this time, regardless of whether they were diagnosed as healthy or with MCI, he reported. On the other hand, people with amyloid all declined at about the same average rate, regardless of diagnosis (see Lim et al., 2014; Lim et al., 2014).
How do the rates of amyloid accumulation and memory loss compare to each other, and to other biomarker change? To relate these very different measures, Maruff calculated the standard deviations of each marker based on the population averages at baseline. He found that in preclinical AD (cognitively normal controls with brain amyloid), amyloid and cognition both worsened by about half a standard deviation over three years. Hippocampal volume shrank only by about one-tenth of a standard deviation. By contrast, in prodromal AD (mild cognitive symptoms and brain amyloid), amyloid, cognition, and hippocampal volume all worsened at the same rate, again half a standard deviation over three years. The acceleration of hippocampal atrophy thus represents a big biomarker difference between the two conditions, Maruff said. He noted that because all three biomarkers change at equivalent rates in prodromal AD, they might all be useful as progression markers in clinical trials.
On the other hand, subjective memory assessments made by patients or caregivers change more slowly, and therefore may be a less-sensitive marker, Maruff added (see Hollands et al., 2015). This finding appears to pour some cold water on recent hopes that subjective memory concerns could be formalized into a useful predictor early on in the symptomatic phase (see Sep 2014 news story). Maruff noted inconsistencies between the AIBL finding that subjective memory may be insensitive to early amyloid-related changes and the findings in other cohorts, for example the Harvard Aging Brain Study, that subjective memory is a sensitive indicator of abnormal amyloid. These, he said, may result from the different ways in which the samples were recruited. Healthy older adults in the AIBL study were recruited from the community and hence may have been less aware of subtle changes in their memory than volunteers recruited from hospital outpatient clinics.
Maruff also clarified that although the average rate of decline is the same in these groups, quite a bit of individual variation occurs. One factor influencing this is ApoE genotype. ApoE4 carriers with amyloid decline faster than non-carriers with amyloid, Maruff said.
Other speakers dug more deeply into the role of ApoE and other modifying factors. For example, Karen Rodrigue of the University of Texas at Dallas had previously reported that ApoE4 carriers with hypertension accumulated amyloid faster than carriers with normal blood pressure, particularly if the hypertension was uncontrolled (see May 2013 news story). That study involved only 118 people, raising the question of whether the findings would hold up in a larger sample.
At CTAD, Rodrigue described her analysis of 1,013 ADNI participants with normal cognition, MCI, or AD. Forty-four percent had at least one copy of ApoE4, and 65 percent had high blood pressure. In agreement with earlier findings, Rodrigue found that people with both risk factors had significantly more amyloid than those who had one or neither. Among the ApoE4 carriers, hypertension nearly doubled the risk of having a positive amyloid scan. In this analysis, a person’s clinical diagnosis did affect the relationship. Specifically, among cognitively normal people, having both risk factors worsened amyloid only if they were over 70. In the MCI group, the interaction between ApoE, hypertension, and amyloid load did not reach statistical significance. In ADNI, the MCI cohort was defined by clinical diagnosis, and biomarker analysis later revealed that many of them did not have brain amyloid. People with AD had the strongest interaction between these factors.
Overall, the data presented at CTAD strengthened the case that amyloid hastens cognitive decline, but also underscored that pure clinical diagnoses poorly capture the underlying pathology. In many current clinical trials, researchers screen potential participants for amyloid pathology or an Aβ/tau CSF signature to ensure that their symptoms are most likely due to Alzheimer’s (see Part 4 of this series).—Madolyn Bowman Rogers.





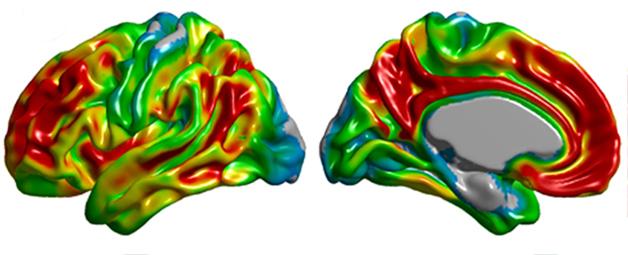
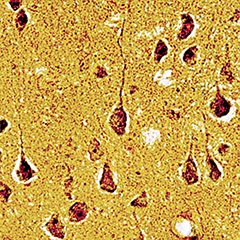



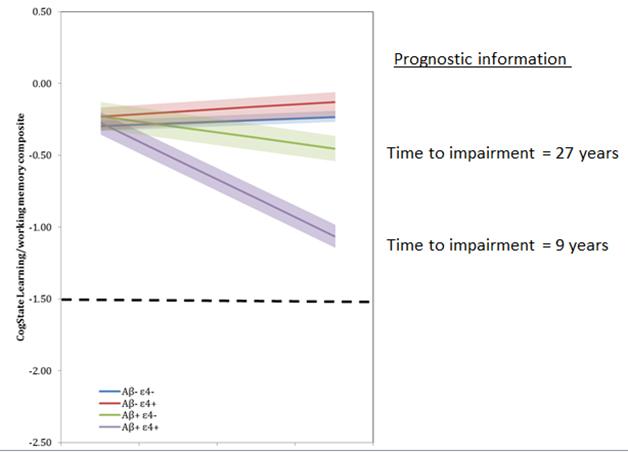
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.