The FDA approval for aducanumab left clinicians with little guidance on how to select the right patients and ensure their safety. At this year’s Alzheimer's Association International Conference, leading clinicians presented appropriate-use recommendations that helped fill the void. These include not prescribing the drug to people with cerebral amyloid angiopathies or people on blood thinners, and monitoring vascular health with frequent MRIs. Clinicians welcomed the guidelines and were hungry for more.
Aducanumab: Will Appropriate-Use Recommendations Speed Uptake?
The Food and Drug Administration’s approval of Biogen and Eisai’s anti-amyloid antibody aducanumab on June 7 left many questions unanswered, including how to use the drug in clinical practice. Since then, a group of Alzheimer’s researchers led by Jeffrey Cummings at the University of Nevada, Las Vegas, have tried to fill the gap. They formulated appropriate-use recommendations (AUR) for identifying, treating, and monitoring patients who would most likely benefit. On July 27, at this year’s Alzheimer’s Association International Conference from July 26–30 in Denver, Cummings and a panel of experts outlined the AUR and addressed questions and concerns in a series of three panel discussions.
Researchers and clinicians, who attended the hybrid-style meeting in person or online, welcomed the guidance, saying it helped clarify an overly broad FDA label. Unlike aducanumab's approval, which incited a storm of criticism, there was no controversy about these AUR. Rather, the AAIC audience seemed starved for this guidance, asking more detailed questions about implementing the treatment than the AUR, or speakers, were able to answer. In each discussion session, questions had to be cut off for lack of time.
“The AUR are timely and necessary. They should help with the intended and safe use of aducanumab,” Ron Petersen at the Mayo Clinic in Rochester, Minnesota, wrote to Alzforum. Mary Sano at Mount Sinai School of Medicine, New York, agreed. “The experts who prepared the AUR have done a great service for practitioners,” she wrote (full comment below).
Published July 20 in the Journal of Prevention of Alzheimer’s Disease, these AUR were based on available clinical trial data. They will be revised as real-life and more trial data come in. It is unclear if any medical organizations such as the American Academy of Neurology or the American Geriatrics Society, both of which had come out against approving aducanumab, will adopt them. At the moment, the AUR are merely guidelines.
Confusion over appropriate use may partly explain why the clinical rollout of the new therapy is slow. And with no guarantee that private insurance companies or Medicare will cover the $56,000 yearly price tag (see Part 5 of this series), several large healthcare providers have announced they will not administer the treatment. Some researchers worry that rural areas and underserved communities will have trouble accessing the drug, widening disparities in U.S. healthcare. Biogen is promoting aducanumab to practitioners and directly to the public, with sessions at AAIC targeted to the former, and a controversial ad campaign aimed at the latter. The long-term effects of all this on the development of other AD drugs, including other immunotherapies, remain unclear (see Part 7 of this series).
Who Qualifies for Treatment?
After a fraught process, the FDA granted aducanumab’s marketing license under the agency’s accelerated approval pathway, based on the idea that robust amyloid removal is reasonably likely to produce a clinical benefit over time (Jun 2021 news). The decision unleashed intense controversy, which has not abated (Jun 2021 news series; Alexander et al., 2021). In particular, clinicians were incensed by the FDA’s label, which initially specified only “Alzheimer’s disease” as a criterion for treatment. The agency later narrowed that to people with mild cognitive impairment or mild dementia due to AD (Jul 8 Endpoints news). Even so, many issues around proper use of the treatment remain unresolved.
To clarify matters, Cummings joined forces with Paul Aisen at the University of Southern California in San Diego, Liana Apostolova at Indiana University School of Medicine in Indianapolis, Alireza Atri at Banner Sun Health Research Institute in Sun City, Arizona, Stephen Salloway at Butler Hospital in Providence, Rhode Island, and Michael Weiner at the University of California, San Francisco. The panel combed through the trial data to define more specific guidelines for how local clinicians should select patients and monitor their treatment.
So what are the AUR? For a start, the panel recommended that clinicians obtain a detailed medical history, test cognition, and perform a thorough neurological exam as first steps in their evaluation of whether the patient before them might benefit from aducanumab. On this, discussion at AAIC emphasized the importance of listening to patient concerns about memory slippage, and paying attention to change in the patient's cognition rather than relying on group-based screening cutoffs that may be inaccurate at an individual level. “Screening tests aren’t always sensitive enough,” Petersen noted. Atri agreed. “You don’t treat a number, you treat an individual,” he said at AAIC. The Alzheimer’s Association has convened a working group to develop step-by-step guidelines for patient screening, Atri added.
Dorene Rentz at Brigham and Women’s Hospital, Boston, said that even with these AUR, primary care physicians may have trouble selecting the right patients for aducanumab. “Referral to specialists, including behavioral neurologists, dementia experts, and neuropsychologists, should be required prior to administration of the drug,” she wrote to Alzforum.
The aducanumab trial population was limited to people between 50 and 85 years of age, whose MMSE was 24 or higher. In clinical practice, both these ranges could be broader, the panel suggested. They put no limits on age, and proposed an MMSE of 21, or MoCA of 17, as the lower limit, noting that these scores are statistically indistinguishable from those used in the trials. Other researchers agree that due to differences in education, some people who score this low may still be at an early stage of amyloid accumulation and able to benefit from treatment. As in the trial population, patients could be taking cholinesterase inhibitors or memantine.
As important as whom to treat is whom not to treat, the AUR say. The panel urged that anyone who has cerebral amyloid angiopathy, a blood clotting disorder, or takes anticoagulants should not be on aducanumab, because they are already at higher risk for the brain swelling and microhemorrhages known as ARIA. Likewise, aducanumab is inappropriate for anyone who has had more than four microhemorrhages, or any larger brain bleed.
The panel also recommends that chronic conditions such as diabetes and cardiovascular disease be stable before prescribing aducanumab. Moreover, physicians should rule out other causes for cognitive impairment, including side effects of medication, folate or vitamin B12 deficiencies, vascular dementia, or normal pressure hydrocephalus.
Above all, before prescribing this amyloid-reducing medication, physicians should confirm patients have amyloid plaques in their brains. Given the high concordance between amyloid PET, CSF Aβ42, and even CSF p-tau, this ascertainment can be done by either modality. Because Aβ42 drops in the CSF before plaques show up on PET scans, the panel recommended that, in cases where both are available and only CSF is abnormal, the physician not prescribe aducanumab at that time and instead follow up with another PET scan in one to three years.
What about other neurologic conditions in which amyloid accumulates? The panel recommended against prescribing aducanumab for dementia with Lewy bodies or Down’s syndrome, noting a lack of information on how the antibody would affect these complex conditions. In contrast, they said its use could be considered for autosomal-dominant AD and atypical forms of the disorder, such as posterior cortical atrophy or logopenic aphasia. Even so, patients and their families should be informed about the lack of data on how aducanumab performs in these specific populations.
Managing ARIA—MRIs Galore
A great concern for clinicians is how to manage ARIA. This side effect occurred in 41 percent of clinical trial participants who received aducanumab, compared to 10 percent of those on placebo. Most of this difference came from brain edema, or ARIA-E. Isolated cases of ARIA-H, or microhemorrhages, were equally common on drug or placebo, according to a poster at AAIC by Biogen's Patrick Burkett. The ARIA-E incidence was 35 percent overall.
Notably, ARIA-E was much more common in APOE4 carriers, with an incidence of 42 percent, compared to 20 percent in noncarriers. The panel recommended that physicians discuss with patients and their families the option of getting genotyped for APOE4, but only proceed with that testing if the additional risk posed by an APOE4 allele would be a factor in the family’s decision-making.
Another wrinkle is that the E4 allele may increase Alzheimer’s risk in African-Americans less than it does in Caucasians (Farrer et al., 1997; Jan 2019 news). At AAIC, Gil Rabinovici of UCSF noted that because of the lack of diversity in aducanumab's clinical trial population, no one knows if race affects the risk-benefit analysis for aducanumab treatment in APOE4 carriers. Sudha Seshadri of Boston University bemoaned the lack of information on how aducanumab affects non-whites. “We should strongly advocate for a Phase 3 trial for these underrepresented groups,” Seshadri said.
You Better Love That Scanner. The AUR recommend four scheduled MRIs over the first year of treatment, plus an extra scan any time ARIA symptoms crop up. [Courtesy of Cummings et al., JPAD.]
The panel recommends that aducanumab monthly dosing and MRI monitoring should be the same for all APOE genotypes. The FDA label specifies that aducanumab be titrated up over six months, with the first two doses being 1 mg/kg, the second two doses 3 mg/kg, and the third two, 6 mg/kg. At the seventh month and thereafter, physicians would infuse the full dose of 10 mg/kg. About half of ARIA-E events take place during that titration window, and 90 percent within the first year, according to Biogen.
With this in mind, the panel recommended MRI scans at baseline, before the fifth dose, before the seventh dose, and at one year. Ideally, all MRIs should be done at the same center with the same scanning parameters. Physicians should also order an MRI any time the patient develops symptoms suggestive of ARIA, such as headache, confusion, or dizziness. About one-quarter of people with ARIA develop such symptoms. The AUR are strictly medical guidelines; they do not address who will pay for all these scans.
The AUR suggest that the response to ARIA depend on its severity. For asymptomatic cases that look mild on MRI scans, dosing can continue unchanged. In the trials, 6 percent of these cases worsened enough to become symptomatic. For symptomatic cases, or for ARIA cases that look moderate or severe on MRI, physicians should stop dosing and repeat the MRI scan monthly. Once the ARIA-E goes away, or ARIA-H stabilizes, dosing can resume. Ninety percent of ARIA cases resolve within five months, according to Biogen, leaving one in 10 unresolved by that time. About 1 percent of clinical trial participants developed severe symptoms; in such cases, the physician should take the patient off aducanumab permanently, the panel said.
“I found the suggested algorithm around ARIA monitoring and management particularly helpful, as outside of trialists few clinicians will have had experience managing this common adverse effect of the drug,” Rabinovici told Alzforum (full comment below). Kejal Kantarci at the Rochester Mayo Clinic thinks even more guidance is needed. “Because MRI is so critical in the monitoring of patients for safety, we may also need technique-based recommendations on how to best monitor with the available technology and give guidance to neuroradiologists,” she wrote to Alzforum.
When to Stop? It’s Anyone’s Guess
On the question of when to stop treatment, aducanumab trial data offer no guidance. According to the AUR, the decision could be based on what the patient or family want, for example if the side effects or monthly injections become too burdensome to continue, or if the treatment does not seem to be helping.
And how would the patient, family, or physician know if it is helping? The panel members suggested that cognitive and functional decline be monitored with easy-to-administer clinical tools, such as the MMSE, MoCA, AD8, the NPI questionnaire, or FAQ. Importantly, cognitive decline is expected to continue even if aducanumab works, and notoriously variable rates of decline in AD will make it challenging to determine if aducanumab is slowing it down, the panel acknowledged. No guidelines are in place to determine if any such slowing exceeds a minimum clinically important difference (Liu et al., 2021). Many researchers have questioned whether the small benefit seen in the aducanumab trials would be detectable to patients.
For his part, Salloway, who was a site P.I. on these trials, believes it is possible to identify people who stay in the mild stage of AD longer than expected. “There are clear responders,” he said at AAIC. Rabinovici suggested that in the future, effectiveness might be judged by the change in biomarkers associated with cognition and synaptic function. In any case, researchers agree that treatment should be stopped when patients progress to moderate AD, as seen by an MMSE below 20 or a CDR of 2 or more.
What to do once aducanumab has cleared all plaques from the brain? Stop giving the drug, as was suggested by the trial of Eli Lilly's anti-amyloid antibody donanemab (Mar 2021 news)? Biogen's data provide too little evidence on this point, the AUR suggest. “Once significant amyloid lowering has been achieved, it may be possible to reduce the frequency of infusions,” the authors noted.
Uptake Slow, Despite Push from Sponsor
With the question of insurance coverage still wide open, few healthcare systems are offering aducanumab just yet. Two large systems have said they will not administer the infusions at this time: the Cleveland Clinic, based in Ohio, and the Mount Sinai Health System in New York (Jul 15 Fierce Healthcare news). Individual physicians in these systems can prescribe the drug for their patients, who then have to go elsewhere to get the infusions. It’s unclear how many clinicians will sign off on aducanumab use. A survey of 200 primary care physicians and neurologists found that two-thirds doubted the drug’s benefits (STAT news).
As of this writing, about 100 patients across the U.S. have begun to get the treatment (STAT news). Marwan Sabbagh at the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas told Alzforum that he has prescribed aducanumab for some patients, but has had to scramble to find places where they can get the infusions. “I’ve gotten names of doctors around the country who are infusing, but it’s been piecemeal,” Sabbagh noted. One such provider is Amber Specialty Pharmacy, which has 21 locations throughout the U.S. (Businesswire). At the moment, patients who want aducanumab may have to be willing to travel for it.
At AAIC, researchers said the limited access is likely to deepen racial and geographic inequities in healthcare. Many people live in “neurology deserts” without access to specialists, an audience member pointed out. Rentz suggested the increasing acceptance of telehealth could help. “Our clinic is open to consultations from around the world. We do cognitive testing over Zoom,” she said.
The other side of the coin is that only about a quarter of AD cases are diagnosed at the MCI stage, and this rate is even lower for minorities. “Brain health needs to be part of the conversation in primary care,” Kate Possin of UCSF said at AAIC.
Meanwhile, Biogen is promoting aducanumab use to physicians and patients. At AAIC, the company sponsored talks on early diagnosis of MCI using biomarkers, and on how to put together multidisciplinary healthcare provider teams to manage diagnosis and treatment. Targeting consumers directly, the company launched a marketing campaign advising people to seek medical advice for mild memory problems (e.g., see website). One ad in The New York Times drew criticism from Madhav Thambisetty at Johns Hopkins University School of Medicine in Baltimore, who noted use of misleading statistics (STAT News).
Sabbagh said that every one of his AD patients asks about aducanumab. “The interest from the patient side is very robust. The demand is there. Physician preparedness is not,” he told Alzforum.
Some researchers see answers to some of these questions forthcoming. “I expect there will be an evolution in our patient selection and management when treating patients with aducanumab, and the recommendations may evolve accordingly,” Kantarci said. Rabinovici agreed. “We are at the beginning of a new era of AD therapy, and it will be exciting to see how we refine our approach to patient care as we gain ‘real world’ experience with molecular therapies,” he wrote.—Madolyn Bowman Rogers
On Donanemab, Plaques Plummet. Off Donanemab, They Stay Away
The FDA’s controversial approval of aducanumab hinged on the premise that clearance of amyloid would be “reasonably likely” to bestow a cognitive benefit. Data presented at the Alzheimer’s Association International Conference (AAIC), held July 26-30 in Denver and online, support the idea that two other antibodies could clear that low bar, as well. Scientists from Eli Lilly reported that the plaque-dissolving strength of donanemab, an antibody trained against forms of Aβ detectable only in plaques, tracked closely with plummeting plasma p-tau217. Weaving their data into a disease-progression model that had been generated from past trial data, they claimed that the amyloid- and tau-lowering effects of the drug correlated with a slowing of cognitive decline. Separately, data from lecanemab’s Phase 2 trial and open-label extension studies provided yet more support for that antibody’s disease-modifying effect, despite the travails that have beset its path through clinical development.
“I was mightily encouraged by the data presented by Lilly and Eisai, which gave further support for the FDA assertion that there was a ‘reasonable likelihood’ that lowering the Aβ-load would result in clinical benefit,” commented Colin Masters of the University of Melbourne, Australia.
Both donanemab and lecanemab have received breakthrough therapy status from the FDA. Similarly to aducanumab, this means that their sponsors could apply for accelerated approval based primarily on changes in surrogate biomarkers that demonstrate amyloid reduction. Both Lilly and Biogen/Eisai have announced plans to use their Phase 2 data to apply for accelerated approval of donanemab and lecanemab, respectively. Lilly will file later this year (see Endpoints News).
“Lilly is in the catbird seat with donanemab,” said Lon Schneider of the University of Southern California in Los Angeles. He noted that Lilly’s Phase 2 trial met its primary endpoint, and its biomarker data showing both amyloid and tau reduction were more convincing than Biogen’s on aducanumab. Even so, Schneider continues to disagree with the new precedent set by the FDA, and favors that Phase 3 clinical efficacy be demonstrated prior to approval of any therapeutic antibody.
Alzforum covered results of the Phase 2 donanemab trial—called TRAILBLAZER—in March, just as the data was also published (Mar 2021 conference news; Mintun et al., 2021). At AAIC, John Sims and Mark Mintun of Lilly reported fresh analyses since then of the amyloid and tau biomarker data, respectively.
The trial enrolled 257 participants who had early symptomatic AD, amyloid in their brains, and—notably—an intermediate level of neurofibrillary tangles based on PET scan. After an initial period, when 131 volunteers randomized to the treatment group gradually received higher and higher doses of donanemab, the trial settled in on monthly infusions of 1,400 mg donanemab. This was given until a person’s amyloid burden dropped below 25 centiloids—the level in healthy young controls—at which point the dose was lowered to 700 mg. If amyloid fell below 11 centiloids, or below 25 for two consecutive scans, the person was switched to placebo.
As previously reported, the 76-week trial met its primary cognitive endpoint, showing a 32 percent slowing of decline on the Integrated Alzheimer’s Disease Rating Scale (iADRS). By 24 weeks, donanemab had completely cleared plaques in 40 percent of participants in the treatment group; by the trial’s end, 68 percent had reached normal levels.
At AAIC, Sims broke down that amyloid reduction data further. Most participants in the treatment group had a rapid reduction in amyloid over the first 24 weeks. As might be expected, the amount of amyloid they shed over those first six months depended upon how much they started with, Sims reported. In other words, people with higher levels of amyloid at baseline lost more than those who started with less. Still, they took longer to dip into the realm of healthy young controls.
Sims reported that once a person’s amyloid complete cleared, their levels stayed down for the remainder of the trial. Among the participants with “deep amyloid clearance,” i.e., amyloid levels below 11 centiloids, and who were switched to placebo by 24 weeks, amyloid burden crept up only slowly by 76 weeks, barely cresting 11 centiloids, on average. At this rate, it would take 14 years for amyloid to accumulate back to baseline level for this group, or about 90 centiloids, Sims reported (see image below).
Get Out, Stay Out. Among people who had complete removal of amyloid plaques by 24 weeks, and were switched to placebo at that time, amyloid burden stayed within the range of healthy young controls (less than 25 centiloids) for the remainder of the trial. [Courtesy of Eli Lilly.]
How did this rapid amyloid clearance influence downstream parts of the AD cascade, i.e., tau tangles and cognitive decline? All participants underwent flortaucipir-PET scans at baseline and 76 weeks. Sims reported that compared to the placebo group, those who had had complete clearance of amyloid by 24 weeks had a significant dip in tangle burden in the temporal, parietal, and frontal lobes by the end of the trial. People who had had partial amyloid clearance by 24 weeks also had significant drops in tau tracer uptake, at least in the parietal and frontal lobes. “These findings link us back to the concept of AD as an amyloid-induced tauopathy,” Sims said.
Would riddance of amyloid also track with slowing of cognitive decline? Among all participants, including those in the placebo group, the percent change in amyloid burden by 24 weeks had no significant correlation with the change in iADRS scores between baseline and 52, 64, and 76 weeks, though there was a trend toward slower cognitive decline among those who had cleared the most amyloid.
Given the small numbers of participants in the Phase 2 trial, Sims further analyzed the trial data using a disease-progression model developed at Pfizer that was based on data from nearly 5,000 controls (Conrado et al., 2014). The Coalition Against Major Diseases had gathered this control data, including from the placebo arms of 15 AD clinical trials conducted between the 1990s and 2010 on people who had mild to moderate AD (Jul 2013 news; Dec 2010 news). The model calculates trajectories of disease progression—as gauged by ADAS-Cog scores—among populations with different characteristics, such as ApoE4 carriers or noncarriers.
Model Comparison. By plugging percent amyloid reduction into the Conrado model, Sims found a statistically significant relationship between amyloid clearance and slowing of disease progression. [Courtesy of Eli Lilly.]
When Sims plugged data from TRAILBLAZER into the model, it estimated that donanemab slowed cognitive decline by 28 percent among all participants in the treatment group, and by 42 percent among APOE4 carriers. The findings jibe with the 32 percent slowing of cognitive decline that the researchers had previously reported, though the model lowered the p value, at less than 0.001. The model also rendered a statistically significant link between amyloid reduction and disease slowing.
Schneider pointed out that the model used data from people who had mild to moderate AD, whereas TRAILBLAZER selected participants with MCI to mild AD. “So there’s a good chance that many of the inputs from the TRAILBLAZER trial are where the model is least accurate,” Schneider said. Still, he and other researchers felt that the Lilly researchers were doing their best with the limited data available.
Plasma p-tau217 Tracks with Amyloid Drop
Mintun’s presentation focused squarely on freshly garnered data from Lilly’s in-house plasma p-tau217 assay. Lilly originally developed the assay on the Meso Scale Discovery platform, which works like an ELISA. At AAIC, Mintun debuted the assay’s adaptation to the more sensitive Simoa platform. Several groups have reported that p-tau217 in the CSF and plasma rise early in the disease trajectory and correlate with amyloid and tangle load (Apr 2020 conference news; Jul 2020 conference news). In a first for the field, Mintun reported how plasma p-tau217 levels changed in response to treatment.
At baseline, plasma p-tau217 correlated with baseline amyloid-PET and tau-PET measures as reported previously. This may seem like a no-brainer, but the result is striking considering that trial enrollment was restricted to people with an intermediate level of tau tracer uptake, thus limiting the dynamic range of PET measurements at baseline, Mintun said.
What happened to p-tau217 as brain amyloid plummeted in response to donanemab treatment? In short, it followed suit. Mintun reported that by 12 weeks, plasma p-tau217 was already significantly lower in the donanemab group compared to placebo. Levels continued to fall at every time point, while statistical significance strengthened. By the end of the trial, plasma p-tau217 had fallen by 24 percent in the treatment group, and risen by 6 percent in the placebo group. The latter rise is on par with that expected during disease progression, Mintun said.
Plasma p-Tau217 Plummets. In response to donanemab treatment, amyloid plaque burden (left) and plasma p-tau217 dropped throughout the trial. [Courtesy of Eli Lilly.]
Notably, at 24, 52, and 76 weeks, the drop in plasma p-tau217 from baseline was strongly tied to reduction in amyloid. In addition, reduction in p-tau217 was linked to reduction in tau tangles, according to tau-PET, between the baseline and the end of the trial.
Mintun also applied the p-tau217 data to the disease progression model that Sims had employed. This cast change in plasma p-tau217 as a significant predictor of slowed disease progression.
Henrik Zetterberg of the University of Gothenburg in Sweden called the responsiveness of plasma p-tau217 to donanemab treatment “a Hallelujah moment,” noting that this was the first time rigorous plasma p-tau biomarker data had been presented in the context of a clinical trial. He said the findings provide strong support for the amyloid cascade hypothesis. That p-tau217 dropped so quickly in response to amyloid removal supports the idea that neurons actively release phospho-tau in response to exposure to amyloid. “If we reduce amyloid exposure, we would expect to see a very quick decrease in phospho-tau secretion,” Zetterberg said. “And that’s exactly what we see.”
This data was also a meeting highlight for Oskar Hansson of Lund University, Sweden. “These important results clearly indicate that p-tau217 in plasma might be used as an easily accessible and cost-effective marker revealing effects of novel treatments on the levels of amyloid-β fibrils in the brain,” Hansson wrote. However, he emphasized that it is not yet known whether plasma p-tau217 can be used as a surrogate biomarker of clinical efficacy. “We simply do not yet know if the reduction of extracellular levels of soluble p-tau is an epiphenomenon merely associated with a reduction in β-amyloid fibrils in the brain, or a key event that will always lead to diminished tau aggregation, less neuronal dysfunction, and reduced degeneration, independently of the mechanism causing the reduced levels of extracellular p-tau to begin with.”
Paul Aisen of the University of Southern California in San Diego made a similar point. “The treatment-related fall in p-tau217 must still be linked to clinical benefit as validation of its utility in guiding therapy,” he wrote. “But if it can be validated, this assay will be an enormously useful tool in the drug-development process.”
David Morgan of Michigan State University in Grand Rapids, who attended the meeting in person, asked Mintun if and when they would measure other blood biomarkers that could hint at a neuroprotective effect of treatment, such as neurofilament light (NfL) and glial fibrillary acidic protein (GFAP). Absent solid cognitive data, perhaps these markers could get closer to addressing the question of efficacy, Morgan told Alzforum. Mintun said that they intend to measure other markers, but optimizing the p-tau217 assay on the sensitive Simoa platform had been top priority leading up to the meeting.
Zetterberg believes this effort may have been well worth it. Given the vanishingly small concentration of p-tau217 in the plasma, the added boost of sensitivity may have been crucial for detecting the treatment effect. Zetterberg said that while he looks forward to seeing how neurodegenerative biomarkers respond to donanemab treatment, the data could prove difficult to interpret. “As microglia are recruited to donanemab-bound plaques, GFAP levels might go wild,” he said. NfL could even rise for a time, he added, as glial cells nosh on detritus from amyloid-exposed neurons. Zetterberg acknowledged that while potentially interesting and important, complex biomarker data might be the last thing Lilly wants while seeking accelerated approval of donanemab by the FDA.
The most exciting implication of the donanemab data, Zetterberg said, was the possibility of treating patients for only a few months to remove plaques. Then, plasma p-tau markers could be used to monitor patients for the return of plaques, at which point they could take another round of treatment, he suggested. Zetterberg was heartened by the data showing that neither Aβ plaques nor plasma p-tau217 appeared to rise within a year after donanemab treatment stopped.
If approved, “amyloid plaque removal” could become a common treatment for people in their golden years who go to their doctors with cognitive complaints, Schneider said.
What about for cognitively normal people who have amyloid plaques? Could donanemab be approved for them? For now, aducanumab is only conditionally approved for people in the early symptomatic stages of AD. Both donanemab and lecanemab trials have included only this population as well. That will soon change, however, when TRAILBLAZER-ALZ3 begins. This Phase 3 trial will test donanemab in cognitively unimpaired people at risk for AD, and will use plasma p-tau217 to select participants (Jul 2021 news).
Lecanemab is also being put to the test in people with preclinical AD in the AHEAD3-45 trial, which is enrolling participants. At AAIC, Chad Swanson of Eisai presented data from the Phase 2 study of lecanemab and an 18-month open label extension.—Jessica Shugart
Antisense Therapy Stifles CSF Tau in Mild Alzheimer’s Disease
While many players in the tau therapy field are banking on immunotherapies to clear neurofibrillary tangles from the brain, some are focused on stopping tau production. In a poster presented at the Alzheimer’s Association International Conference, held July 26-30 in Denver and online, Catherine Mummery, University College London, Candice Junge, Ionis Pharmaceuticals, Carlsbad, California, and colleagues gave a peek at data from the first-ever trial of a tau antisense oligonucleotide in mild Alzheimer’s disease. In this Phase 1b trial, BIIB080, developed by Ionis in partnership with Biogen Inc., Cambridge, Massachusetts, caused no serious adverse events. It did reduce both total tau and phosphotau-181 in the cerebrospinal fluid by 30 to 50 percent. What this means for cognition remains unclear. A Phase 2 trial is planned to begin in mid-2022.
“This is a landmark study of human tau therapeutics,” Adam Boxer, University of California, San Francisco, told Alzforum. “It is the first to target tau at the genetic level and shows that, in a small number of people for a relatively limited duration, a significant reduction of tau protein in CSF is safe and well-tolerated.” Gil Rabinovici, also at UCSF, agreed, noting that “ASOs are a very promising potential approach to treating tauopathies.”
Antisense oligonucleotides match to specific snippets of mRNA, suppressing translation. They have seen a revival, spurred in part by the approval of Ionis’ nursinersen for spinal muscular atrophy (Nov 2016 news; May 2018 conference news). ASOs are being tested for their potential as therapeutics in neurodegenerative diseases, including AD (Aug 2019 news).
For this Phase 1b study, researchers recruited 46 people ages 50 to 74 with mild AD from the U.K., Canada, Germany, Sweden, Finland, and the Netherlands. AD diagnosis was determined by an overall Clinical Dementia Rating score of 1, Mini Mental State Exam score of 20 to 27, and CSF amyloid positivity.
Participants were randomized 1:3 to receive placebo or BIIB080 at low, medium, and high doses. Researchers administered drug or placebo via intrathecal injection every four weeks for three months, followed by a six-month monitoring period (see image below). One group received two high doses three months apart. Once the monitoring period wrapped up, everyone began receiving the ASO at his or her assigned dose every 13 weeks for another year with up to six months of monitoring afterwards. This open-label extension (OLE) is expected to wrap up in early 2022.
Dosing and Testing. Participants received low (cohort A), medium (cohort B), and high (cohorts C and D) doses of BIIB080 over a three-month treatment period. That was followed by a six-month sampling period. [Courtesy of Catherine Mummery, University of College London.]
The primary outcome measure was adverse events, while secondary outcomes included CSF tau and pharmacokinetics. No participants had serious adverse events and 43 have completed the post-OLE monitoring. Three-quarters of the placebo group reported at least one side effect, as did 94 percent of those who received the drug. Up to 30 percent of the treatment group experienced mild to moderate effects, such as headache, pain while the drug was being injected, or post-lumbar puncture syndrome (see image below). The latter occurs if the punctured dura mater leaks CSF, dropping intracranial pressure and causing a characteristic posture-dependent headache (Cohen et al., 1991).
Painful for Some. A majority of participants in both placebo and treatment groups reported mild side effects, which the researchers attributed to the lumbar puncture procedure itself. [Courtesy of Catherine Mummery, University College London.]
Did the drug hit its target? CSF total tau and phospho-tau181 levels fell over time, dropping up to 35 percent in the low-dose group and up to 50 percent in the high-dose group eight weeks after the last dose (see image below). In people who received the high dose, the markers continued dropping over the next eight weeks, falling another 5 percent.
Other tau immunotherapies currently in trials, such as anti-tau antibodies, focus on extracellular tau, nipping its spread and aggregation in the bud. In contrast, ASOs go after expression, which can quash tau levels inside and outside of cells. “Knocking down all tau is a more exciting and logical approach to me, given our uncertainty about which isoform is pathological,” Mummery told Alzforum. Boxer agreed. “Even if the hypothesis that prion-like spread is important for tauopathies is not correct, genetically suppressing tau with ASOs will give us a much better idea of whether tau is central to neurodegeneration,” he told Alzforum. Rabinovici noted that slashing all tau may make this approach applicable to multiple tauopathies, since each involves different oligomers and fibril conformations.
Tumbling Tau. Both total tau (top) and p-tau181 (bottom) dropped in the CSF of participants treated with low (yellow), medium (red), or high (blue and purple) dose BIIB080, while tau in people given placebo (black) remained unchanged. Arrowheads indicate when the drug or placebo were given. [Courtesy of Catherine Mummery, University of College London.]
But what do lower CSF tau levels mean clinically? For now, we do not know. “The strong dose relationship shows a real biological effect, but the big question is whether that translates into clinical benefits,” Mummery told Alzforum. The anti-tau antibodies semorinemab and gosuranemab also lowered tau in the CSF but had no clinical benefit in AD (May 2021 news; Jun 2021 news).
Ditto for Ionis’ ASO for Huntington’s disease. Tominersen, also called HTTRx, halved mutant Htt levels in CSF, but provided no clinical improvement for patients. A Phase 3 trial screeched to a halt this March (Mar 2018 news; company press release).
“The pathological mechanisms are unique among neurodegenerative disorders, so it is difficult to compare ASOs across indications,” Junge wrote to Alzforum. Rabinovici agreed. “The success of one ASO may, or may not, predict the success of others,” he said.
Although it is still early days, Boxer is hopeful for BIIB080, noting that when it lowered tau in the CSF of nonhuman primates, levels of tau fell in their hippocampi as well, as judged by postmortem analysis (Jan 2017 news; DeVos et al., 2017). Mummery agreed. “The quality of the preclinical work is one of the reasons I am very excited about this drug's potential,” she said.
Eric Reiman, Banner Alzheimer’s Institute, Phoenix, wondered how the ASO affects other fluid markers. “Additional biomarker data, including for downstream neurodegenerative biomarkers such as NfL, and clinical data will be needed to clarify the treatment’s disease-slowing effects,” he wrote to Alzforum (full comment below).
Rabinovici raised a concern about how reducing tau low enough to see pathological changes relates to cognition. “With BACE inhibitors, we did not know that aggressive inhibition would hasten cognitive decline until late-stage trials, so how much tau lowering is too much?” he asked.
A Phase 2 trial of BIIB080 for mild AD is planned. “While I haven’t seen the protocol yet, trial sites have been approached to participate and we hope it will begin by mid-2022,” Mummery told Alzforum. She believes the trial will include U.S. and European sites.—Chelsea Weidman Burke
Seeking Real-World Data on Whether Aducanumab Works
The Food and Drug Administration’s conditional approval of aducanumab (Aduhelm) left most researchers and clinicians with a big question: Does the drug really work to slow cognitive and clinical decline? Post-market research will now try to answer this question. At the Alzheimer’s Association International Conference, held July 26-30 in Denver, James Galvin of the University of Miami described one such effort. Galvin introduced an observational study, sponsored by Biogen, that aims to enroll 6,000 people taking aducanumab. It will make a particular effort to recruit minorities, who were under-represented in the clinical trials. The observational study will track the clinical outcomes, blood biomarkers, and safety of aducanumab use. It has no control group.
The study is likely to be one of many attempts to determine aducanumab's real-world effects. Researchers have proposed other models for testing how well the drug works, but given the difficulty of finding a true control group now that treatment is approved, some wonder if the field will ever get a definitive answer. Nonetheless, data from such post-market studies will likely influence payers’ coverage decisions (see Part 5 of this series).
This study is separate from the FDA-required Phase 4 trial, on which no information has been made public yet.
ICARE-AD Will Assess Real-World Aducanumab Treatment
At AAIC, Galvin laid out the details of the International Collaboration for Real-World Evidence in Alzheimer’s Disease, suggestively collapsed into the acronym ICARE-AD. Biogen began identifying potential centers for this study before receiving FDA approval for aducanumab. More than 200 are now part of the effort; Galvin said they include academic and memory clinics, but not primary care. Biogen plans to expand the study to other countries if aducanumab becomes available elsewhere. The contract research organization IQVIA, based in Durham, North Carolina, will run the study. ICARE-AD does not have a principal investigator but will be led by a steering committee co-chaired by Galvin.
Participating clinics will recruit patients over the next four years. Of the 6,000 planned enrollees, the goal is that at least 500 be African-American and 500 Latino. This is an effort to remedy the lack of diversity in the aducanumab Phase 3 trials. Galvin said many of the sites for this study are in areas with diverse populations, which should aid in recruiting minorities. For example, Galvin's clinic in Miami serves a local population that is 60 percent Latino.
Importantly, ICARE-AD will relax the inclusion/exclusion criteria typically used in AD placebo-controlled trials, and include more people with chronic medical conditions such as diabetes, heart disease, and cancer. The long list of usual AD trial exclusion criteria is one reason a disproportionate number of minority participants tend to get screened out of participation (Raman et al., 2021). The idea is for ICARE-AD to reflect the U.S. clinic population, Galvin said. This means the ICARE-AD population may be less healthy overall than the ENGAGE/EMERGE populations were. Clinical trials usually try to select participants with relatively "pure" forms of the disease in question who are otherwise in good health.
ICARE-AD will follow each participant for up to five years, collecting data via interviews roughly every six months. Biogen has selected tests it believes will be both easy to administer and reflect change in early stage AD. The primary cognitive measure will be the MoCA version 8.1; the primary functional measure, the Amsterdam Instrumental Activities of Daily Living Questionnaire Short Version; and the main behavioral measure, the Neuropsychiatric Inventory Questionnaire. To assess global functioning, clinicians will administer the CDR at baseline and will track participants with the Quick Dementia Rating System. In addition, centers will use the Functional Activities Questionnaire and Geriatric Depression Scale Short Form.
Because the study has no control arm, there is no built-in way to directly assess how patients would have fared without going on aducanumab. To get at this question, researchers could compare the ICARE-AD treatment outcome data to historical norms, for example from observational studies such as ADNI, AIBL, BSLA or other biomarker-enhanced aging cohorts. Better yet, scientists could use existing disease progression models built from placebo group data (e.g. Jul 2013 news; Conrado et al., 2014). Lilly uses such a disease-progression model to better understand donanemab treatment data (Aug 2021 conference news). DIAN uses one, as well (Apr 2020 conference news). Galvin indicated to Alzforum that he knows of no plans to compare the ICARE-AD outcome data to anything.
The ICARE-AD study will assess participants' quality of life using the Quality of Life–Alzheimer’s Disease scale and the Short Form–12 Health Survey. It will also track caregiver burden and the cost of dementia care; for these outcomes, the measures are the Zarit Burden Interview and the Resource Utilization in Dementia Lite questionnaire, respectively.
Biogen will bank plasma and serum from participants to measure biomarkers, though as of this writing, Galvin did not know which markers would be chosen, nor whether samples would be collected from all participants, or from a subset. In addition, clinicians will track the incidence and severity of adverse events, particularly ARIA. At AAIC, an attendee asked how MRIs would be standardized across centers. Galvin said no attempt will be made to do this; the data will reflect real-world use.
The audience questioned how feasible this study will be, given the current lack of insurance coverage not only for aducanumab, but also for amyloid screening and for the numerous MRI scans to monitor ARIA. Although Biogen will pay physicians for administering outcome measures, and will cover the cost of CSF analysis for patients interested in aducanumab, Biogen will not pay for any other testing, nor the cost of the drug itself. Galvin suggested that amyloid PET scans could be covered under the new IDEAS study instead, i.e., indirectly via Medicare funds (Aug 2020 conference news). Galvin acknowledged, however, that due to the high cost of aducanumab, ICARE-AD will likely get off the ground only after there is more clarity about insurance coverage (Fierce Pharma story).
Between a delayed ramp-up, recruitment, and observation, ICARE-AD will likely take up to 10 years to generate results.
ICARE will not assess aducanumab’s efficacy. Galvin emphasized the definitional distinction between efficacy, which compares effects between people on and off an investigational drug, and effectiveness, which simply measures the effects of a drug in those who take it. ICARE-AD will track effectiveness. To settle questions of efficacy, clinicians will need a placebo-controlled trial.
So What About Efficacy?
The FDA requires Biogen to complete such a trial, and Biogen has promised to do so sooner than the nine years the agency gave it. Thus far, the company has revealed no details.
In the meantime, other researchers have suggested ways to approximate aducanumab’s efficacy. Maria Glymour at the University of California, San Francisco, proposed that researchers exploit the fact that manufacturing delays and logistical challenges mean aducanumab treatment will begin at different times in different places. Researchers could use these staggered start times to perform a quasi-randomized trial, comparing how quickly patients at different clinics progress to mild or moderate dementia as a function of their total exposure to aducanumab. This model would require clinics to coordinate their efforts, whereby they record run-in data for each patient in the months before aducanumab became available for them. Researchers could then compare that progression data to data from people on drug.
“This study design would provide patients and providers with vastly better information than currently available,” Glymour noted in her proposal.
Madhav Thambisetty at Johns Hopkins University School of Medicine in Baltimore endorsed Glymour's idea. He noted that it could produce answers much sooner than Biogen’s Phase 4 trial will. He said aducanumab's accelerated approval based on biomarkers means that the thousands of AD patients who will eventually take the drug are in effect part of a huge public health experiment to determine whether aducanumab helps on the outcomes that matter to patients. Given this, Thambisetty suggested that medical centers collaborate to collect data on how their patients fare on this drug. “[Patients’] active participation as citizen-scientists in these post-approval studies is now critical to definitively test whether this medication works or not,” Thambisetty wrote (Endpoints News editorial).
Other analysts, such as Rita Rubin at the American Medical Association, warn that the unusual approval process means clinicians may never get a clear answer on how well aducanumab works. Now that aducanumab is publicly available, who will want to enter a randomized controlled trial where they may get placebo (Rubin, 2021)? Accelerated approval “has too often become essentially a commercial end-run around the [FDA],” wrote Elisabeth Rosenthal of Kaiser Health News (Washington Post editorial).
Biogen has not published the data from its two Phase 3 trials of aducanumab, which were conducted from 2015 to 2019. The company recently withdrew a manuscript describing the data from JAMA after the journal asked for edits (Axios news). On August 12, Biogen is publishing, in Nature Medicine, the data from the trials of its discontinued anti-tau antibody gosuranemab, which were conducted from 2017 to 2019 (Dam et al., 2021).—Madolyn Bowman Rogers
The Food and Drug Administration’s controversial June 7 approval of aducanumab (Aduhelm) means that for the first time in 18 years, Alzheimer’s patients have a new treatment option. Nonetheless, due to the high cost of the drug and associated testing, few people will be able to get the treatment unless insurance companies cover it. Whether that will happen remains uncertain, given that aducanumab was approved based on a biomarker effect, without convincing evidence of efficacy. Many big insurers, including the Centers for Medicare and Medicaid Services, are currently weighing their options and seeking input. In private and public meetings, insurers have been consulting with Alzheimer’s researchers, health care analysts, and advocates to glean more information about the drug’s efficacy and safety. Their eventual decisions could determine how widely used aducanumab becomes.
“Many institutions are waiting to hear how reimbursement will be arranged; once that is solved, I think there will be a substantial uptick in use,” Jeffrey Cummings at the University of Nevada, Las Vegas, told Alzforum. Cummings led the expert panel that devised appropriate-use recommendations for aducanumab (Part 1 of this series). Meanwhile, controversy over the approval process for aducanumab continues to brew (see Part 8 of this series).
Payers Are Weighing Options
The uncertainty over efficacy, plus the drug’s $56,000 per year price tag, has left insurers scrambling. Some affiliates of Blue Cross and Blue Shield, and Massachusetts’ Point32Health, have already said they won’t cover the treatment (STAT news; Boston Globe). The Department of Veterans Affairs announced August 11 that it will not carry aducanumab in its formulary, though local VA centers can request access to the drug. The VA recommends against prescribing aducanumab, except in “highly selected patients” who are receiving care at centers with the requisite expertise (Endpoints News). Most insurers are still deciding (Axios news; STAT news).
To help inform their decision, the Margolis Center for Health Policy at Duke University on July 14 convened representatives from about a dozen insurance companies including CMS, plus Alzheimer’s researchers, Biogen staff, and the FDA for a private meeting. It gave insurers a chance to go over the trial data and ask questions about the patient population, efficacy, and safety. Payers were particularly interested in the costs of monitoring for ARIA by MRI, noted Eric Siemers of Siemers Integration LLC, who attended the meeting. The appropriate use guidelines presented at AAIC recommend more MRIs than the FDA label specifies.
“Since this is the first disease-modifying drug with any sort of approval for AD, payers will need time to understand the financial implications,” Siemers wrote to Alzforum. Cummings, who also attended, agreed. “The meeting provided a neutral setting for dialogue. No consensus was reached, but this was a good beginning,” Cummings said.
Private payers are likely to take their cues from CMS. An estimated 80 percent of the patients eligible for aducanumab are covered by Medicare. To establish a consistent nation-wide policy, CMS launched a National Coverage Determination for aducanumab July 12 (CMS announcement; Endpoints News). The agency uses this process for only a few drugs each year. The NCD began with a 30-day public comment period and two public listening sessions. CMS will issue a draft coverage proposal after six months, and will encourage public comments on that proposal for another 30 days. A final decision is expected after nine months, in April 2022.
Of note, CMS has indicated that its decision will cover other anti-amyloid monoclonal antibodies, as well. This has sparked pushback from Eli Lilly, which is filing for accelerated approval for its own anti-amyloid antibody, donanemab. In a public comment, a representative for Lilly urged CMS to consider the merits of each immunotherapy separately (Endpoints News).
In the meantime, decisions about whether to cover aducanumab are up to regional Medicare centers. “Right now, most are saying no,” noted Mark McClellan, who was FDA commissioner from 2002 to 2004 and now runs the Duke-Margolis Center that hosted the private meeting.
One possible outcome for the NCD would be for the agency to grant Coverage with Evidence Determination (CED), the same process currently used to cover amyloid PET scans in the IDEAS study. In this scenario, treatment would be covered as part of a clinical trial that gathers evidence of aducanumab’s effectiveness. McClellan helped develop the CED process while at CMS. He noted that such trials are usually not randomized. Instead, they may match people who decide to get the treatment versus those who don’t, or compare different dosages and timing of treatment.
McClellan lamented that although the FDA has become more open to innovative trial designs, there has been no comparable innovation for figuring out how to cover expensive, groundbreaking therapies. “I hope [aducanumab] is a wake-up call. More innovative treatments are coming,” McClellan said at a July 15 meeting of the Institute for Clinical and Economic Review (ICER).
ICER Doubles Down: Aducanumab Not Worth the Cost
The purpose of the day-long ICER meeting was to review efficacy data for aducanumab and debate whether the treatment is cost-effective. Besides ICER scientists, representatives from Biogen, advocacy groups, and insurance companies attended, along with 15 members of the California Technology Assessment Forum. This forum included an academic geriatrician, neurologist, several other physicians, health policy and economics experts, an ethicist, and three patient advocates. Also attending the ICER meeting were two Alzheimer’s researchers, one of whom, Sarah Kremen of Cedars-Sinai Medical Center, Los Angeles, was a site PI for the aducanumab trials.
In discussion, a sharp division of opinion became evident. On one side, representatives from Biogen, the Alzheimer’s Association, and Us Against Alzheimer’s, as well as a patient and caregiver affected by AD, spoke in favor of aducanumab. On the other side, independent experts from ICER and CTAF were unanimous in finding the efficacy data lacking. Kremen struck something of a middle ground, noting that some patients may benefit from aducanumab, but it would be challenging to determine who those could be, and whether the benefit would outweigh the health risks from ARIA. “From the standpoint of ‘do no harm,’ prescribing makes us uneasy,” Kremen noted in the discussion.
Across the board, participants expressed intense concern over aducanumab’s cost. ICER president Steve Pearson noted that 21 million Americans are in medical debt, and aducanumab is likely to exacerbate this. The drug's estimated $11,300 yearly out-of-pocket cost for Medicare beneficiaries would amount to 40 percent of the median income for a retiree. Six million Medicare beneficiaries lack supplemental insurance, and for those who have it, the out-of-pocket maximum would still be $7,500 per year, Pearson said.
At the ICER conference, the drug’s price was the one issue where ICER and the Alzheimer Association agreed. “We believe the price will hinder access,” said Matthew Baumgart of the association.
Notwithstanding the cost to individuals, the total price tag could reach $57 billion per year if 1 million patients go on drug. That would drain CMS coffers. Biogen's Chris Leibman defended the price, arguing that Biogen expects no more than 10 percent of the roughly one to 2 million eligible patients, or around 150,000 people, to take aducanumab. “If we see different numbers, we will work with CMS and private payers to address pricing,” Leibman said.
An insurance industry analyst noted that patients demand the drug. “We’ve never had more calls from subscribers on day one than we did for aducanumab,” said Leslie Fish of IPD Analytics. The demand makes payers worry that it will be challenging to restrict coverage to the right patient group. “Almost all payers want to set prior authorization,” Fish noted. This would allow insurers to judge the medical necessity and appropriateness of treatment on a case-by-case basis.
Other insurance representatives pushed for value-based pricing. “We’d like patients to have access to it, and we’ll work on a fair price,” said Pat Gleason of Prime Therapeutics, based in Eagan, Minnesota.
Sei Lee of the University of California, San Francisco, summed up the feelings of many. “For extraordinarily high cost, I would expect extraordinary evidence [of efficacy], and I haven’t seen that,” Lee said.
The ICER meeting concluded by having the CTAF members vote on whether the efficacy data was sufficient to conclude that treatment with aducanumab was better than the standard of care. Unanimously, they said it was not. Panelists also voted to say the treatment was likely to do more harm than good, due to its high cost and risk of ARIA.
ICER's Pearson, and many CTAF members, spoke about their own loved ones having suffered from Alzheimer’s, or having the disease now. Some currently take care of an affected parent. They understand the urgency to find better treatments. Patient advocate Richard Seiden said, “I’m optimistic that research will produce better disease-modifying drugs, and I trust clinicians will appropriately manage the expectations of patients and caregivers [around aducanumab].”
In a final report issued August 5, ICER reaffirmed and elaborated its May 5 interim conclusion that aducanumab is not cost-effective (Endpoints News; Endpoints News). Pearson called on Biogen to lower the price. Given that more anti-amyloid antibodies are coming, the broader question of how efficacious this drug, and its class, is remains open. “ICER will come back to this topic,” he promised.—Madolyn Bowman Rogers
Lecanemab Post Hoc: Is Continual Treatment Required for Cognitive Benefit?
Once again, Aβ immunotherapy was the topic du jour at this year’s Alzheimer’s Association International Conference (AAIC), held July 26-30 in Denver and online. Drug companies are moving to Phase 3 studies while simultaneously wooing the FDA for accelerated approval. Among their proffers is lecanemab, an antibody trained against protofibrillar forms of Aβ. After a complicated Bayesian Phase 2 study hinted at efficacy in people with early AD, and participants had been off lecanemab for nine months to five years, Eisai began an open-label extension (OLE).
At AAIC, Chad Swanson, who directs the company’s neuroscience clinical development group in Woodcliff Lake, New Jersey, reported 18-month biomarker and cognitive data from this small OLE, and post hoc analyses across the entire span of the study. He reported that cognition declined more slowly in people on lecanemab than in controls. While decline regained speed during time off drug, people who had received treatment still maintained their edge over those who had been in the placebo group. Open-label treatment slowed cognitive decline once again. The plasma ratio of Aβ42/40 gradually fell during the gap between the blinded and open-label trials—indicating a rise in brain amyloid deposition—but rebounded when open-label treatment resumed. Together, the findings hint at a disease-modifying effect, claimed Swanson. Whether continued dosing would be necessary to maintain a cognitive benefit remains unsettled.
Lecanemab’s Rocky Road
This antibody's path through development has been bumpy. Eisai employed an ambitious Bayesian design for its 856-participant Phase 2 trial. In this type of trial, data are continually measured and enrollees are increasingly randomized to the more-effective doses as the trial progresses. The 18-month trial started with five dose groups and a placebo group. Although the trial missed its 12-month cognitive endpoint, data at 18 months indicated that the highest dose—10 mg/kg bi-weekly—had lowered amyloid burden by 93 percent and slowed, but did not halt, cognitive decline (Jul 2018 conference news).
This dose slowed worsening on the ADCOMS by 30 percent, and nearly halved decline on the ADAS-Cog. That’s similar to donanemab, which docked decline on the iADRS, a cognitive and functional composite measure, by 32 percent in Phase 2. Aducanumab slowed decline on the ADAS-Cog by a quarter in the Phase 3 EMERGE, but had no benefit in ENGAGE (Aug 2021 conference news; Dec 2019 conference news).
The trouble for lecanemab: an uneven distribution of ApoE4 carriers among the dose groups muddled interpretation of the data for a time (Nov 2018 conference news). This kink sorted out, Eisai started the OLE, inviting those who had completed the placebo-controlled, double-blinded “core” portion of the trial to now take the highest, 10 mg/kg twice-monthly dose for another five years. Researchers previously reported that during the gap period between the placebo-controlled and extension—which ranged from nine to 59 months—cognition declined at a similar rate among all groups, although those who had received either of the two highest doses of lecanemab maintained their edge over the placebo group (Dec 2019 conference news). Their amyloid burden also remained low. The data indicated that while plaques had remained at bay during the gap period, cognition had started to wane again, suggesting that continued treatment, perhaps to remove Aβ protofibrils, may be necessary for an enduring cognitive effect.
Further, at last year’s CTAD, Swanson reported amyloid-PET data as far as 12 months into the OLE (Nov 2020 conference news). Among participants who had been on placebo in the trial's core portion and were therefore getting lecanemab for the first time, amyloid dropped to a similar degree as it had among participants treated in the core trial.
At AAIC, then, Swanson reported 18-month cognitive and biomarker data from the OLE. In toto, 180 participants enrolled. At baseline, 24 scored greater than 1 on the clinical dementia rating (CDR) scale, meaning that they had progressed beyond the realm of early AD. The core study had enrolled people with MCI or mild AD (CDR 0.5-1), meaning that these 24 no longer met inclusion criteria for the core trial. Despite this, these progressors, now with moderate to severe AD, continued to receive treatment in the extension.
Swanson presented retrospective data on them. Showing pooled data from people who had been in the placebo and treatment groups, he reported that these 24 progressors had also declined at a more rapid pace on other measures of cognition, namely the ADCOMS, CDR-SB, and ADAS-Cog, during the core and gap periods, compared to the 147 participants who were still at the early AD stage by the beginning of the OLE. Swanson did not break down the 24 progressors by treatment group in his presentation, but he told Alzforum that lecanemab treatment did not influence their rate of cognitive decline, either in the core or later in the extension. Essentially, these participants did not respond to lecanemab treatment. Swanson said that efforts are underway to investigate whether baseline biomarkers, including tau PET or fluid biomarkers, could have identified this group of non-responders from the beginning of the trial.
Non-Responders Identified. Of 180 people in the open-label extension, the 24 whose global CDR was 1 or higher at baseline (red line) had declined more rapidly on ADCOMS during the core (blue) and gap (orange) periods of the trial than the people who started the OLE with milder dementia (blue line). Pink section reflects a three-month safety follow-up. [Image courtesy of Biogen/Eisai.]
Swanson excluded the 24 fast progressors from subsequent analyses. Among the remaining participants, those who had been in the placebo group from the get-go had worsened during the core trial on ADCOMS at a similar rate as did people with mild AD in the ADNI cohort. On the other hand, people in the two treatment groups had worsened at a slower rate that those historical controls. Moving on to the gap period, Swanson showed that decline continued apace among all three groups, with the placebo group continuing to do worse, and the lecanemab groups doing better than ADNI controls. This suggests an enduring disease-modifying effect, Swanson said. These progression rates in the gap period were based on measures taken at a single time point at the beginning of the OLE; the gap period varied widely.
What happened during the extension itself? Swanson presented data from a subset of 62 people who had completed 18-month cognitive assessments in the OLE, and had been in the placebo, 10 mg/kg monthly, or 10 mg/kg biweekly groups during the core. This group excluded the fast progressors. Measuring change in ADCOMS relative to OLE baseline, Swanson found that while the people who had previously been on placebo and switched to high-dose lecanemab initially declined like ADNI controls, lo and behold, this worsening slowed after six months and plateaued through 18 months, suggesting the immunotherapy was having an effect (see image below).
Stable in Open Label? In the OLE, everyone took the highest dose of lecanemab. People who had been on placebo (black) or the highest dose (green) in the core study, declined more slowly on ADCOMS after six months, while those who had been on the second-highest dose in the core (blue) continued to decline, though not as fast as ADNI controls (orange). People in the second-highest dose group in the core trial were more likely to be ApoE4 carriers. [Image courtesy of Biogen/Eisai.]
The drug similarly altered the rate of cognitive decline in people who had previously taken the highest dose of the drug, showing stabilization after six months. However, those in the second-highest dose group behaved more like ADNI controls, continuing to decline over the 18 months. Notably, this group had a higher proportion of ApoE4 carriers than the other two groups.
Swanson showed no absolute scores, only change from OLE baseline. The data were also based on small numbers of participants, and Swanson emphasized that the primary purpose of the analysis was to generate hypotheses that could be tested in the ongoing Phase 3 study, CLARITY-AD.
How did lecanemab influence Aβ accumulation throughout the trial? Swanson had shown previously that the highest dose of lecanemab had lowered plaque burden by more than 90 percent, as per PET, and that amyloid deposits had largely remained at bay during the gap period. At AAIC, he offered more support for that finding, reporting ratios of Aβ42 to Aβ40 in the plasma, which had been collected in both core and extension periods. As more Aβ42 gets released from fibrils and plaque decorated with antibodies, scientists expect the plasma level to rise. In keeping with this, plasma Aβ42/40 held steady in the placebo group during the trial's core period, but rose in the two high-dose treatment groups. Between the end of the core period and the beginning of the OLE, the ratio had crept downward in the treatment groups, suggesting a surge of amyloid deposition off drug. Even so, the ratio remained well above that in placebo (see image below).
Six months into the extension, the Aβ42/40 ratio of the people who had switched from placebo to high dose had nearly caught up with those of the two previously treated groups, suggesting a rapid cessation of amyloid deposition.
Shifty Plasma. During the trial's core period (blue), the ratio of Aβ42/Aβ40 increased in the two treatment groups (green, blue lines) but held steady in the placebo group (black line). The ratio fell between the core and OLE (yellow), then rose again once treatment resumed. [Courtesy of Eisai.]
“While the clinical treatment effects in the OLE are difficult to evaluate with very small numbers and absent a control group, the movement of the plasma Aβ42/40 ratio off and on treatment is potentially of considerable importance and merits significantly further investigation and replication,” wrote Howard Feldman of the University of California, San Diego, to Alzforum.
To Swanson, the Aβ42/40 data suggest lecanemab effectively stops Aβ accumulation. However, once treatment ends, Aβ starts accumulating again, even if plaques are not yet detectable. He interpreted both the biomarker and cognitive findings as evidence that continued treatment with lecanemab may be necessary to keep Aβ build-up, and cognitive deterioration, at bay.
David Morgan of the Michigan State University in Grand Rapids interpreted the data differently. He noted that while plasma Aβ42/40 did dip during the gap period, it remained well above the placebo group. He made a similar point about the cognitive data, noting that the differences between placebo and treatment groups were maintained throughout the gap period. To his mind, this implies a continued disease-modifying effect in the absence of treatment. By comparison, in a Phase 2 trial of donanemab, plaques dropped to the level of healthy controls, and were still low a year after treatment was stopped (see Part 2 of this series). Cognitive benefit still persisted after treatment had stopped, as well.
Henrik Zetterberg of the University of Gothenberg in Sweden hesitated interpreting the plasma Aβ42/40 findings. He noted that in previous trials of Aβ monoclonals, the antibodies stabilized Aβ in the plasma, thus increasing the peptide's half-life there. Without data on the absolute concentrations of Aβ42 and Aβ40, it is unclear whether the plasma ratio truly reflects changes in Aβ accumulation in the brain, he cautioned.
In addition to the ongoing extension study, two Phase 3 studies are putting lecanemab to the test. In March 2019, CLARITY-AD started enrolling people with mild AD into an 18-month early AD trial, whose primary outcome is change on the CDR-SB. At AAIC, Eisai's Michelle Gee showed the baseline characteristics of participants enrolled thus far. Of more than 1,700 projected participants, 1,536 have been selected at sites in North America, Europe, Asia, and Australia. For the most part, the randomized population seems very like the participants in the Phase 2 study, with an average age of 72 and cognitive scores indicative of early AD. Enrollment is complete in all countries bar China, where it continues. Preliminary results are expected in September 2022.
The more recent AHEAD 3-45 secondary prevention study is seeking cognitively normal people with signs of amyloid in the brain (Nov 2020 conference news). AHEAD 3-45 combines two studies, A3 and A45. A3 is evaluating lecanemab's ability to lower plaque in people with only 20 to 40 centiloids of amyloid, and has a primary endpoint of amyloid reduction. The Phase 3 A45 enrolls people with intermediate levels of amyloid—above 40 centiloids—and uses a cognitive endpoint, the PACC-5.
At AAIC, Jin Zhou of Eisai reported that this four-year trial, run by Eisai and the Alzheimer’s Clinical Trial Consortium (ACTC) has so far screened 881 people. Of those, 209 were excluded prior to receiving an amyloid-PET scan, based on general health or cognition. Of the remaining 672 who received an amyloid-PET scan, 297 were excluded because their amyloid levels were too low, leaving only one third eligible for the trial. Amyloid-PET was by far the most common reason for exclusion. While a computational simulation based on ADNI data had projected that about half of screened participants would meet the amyloid-PET selection criteria, in reality only 32 percent have, so far. As of June 30, 2021, 77 people have been randomized—23 people had enrolled in A3, and 54 in A45, Zhou reported.
Even as the Phase 3 trials push ahead, Biogen/Eisai recently announced plans to apply for accelerated approval of lecanemab (Endpoint News), following in Eli Lilly’s footsteps with donanemab.—Jessica Shugart
How will aducanumab’s approval influence people’s willingness to join, and stay in, future Alzheimer’s disease clinical trials? At the Alzheimer’s Association International Conference, held July 26-30 in Denver and online, researchers said the effect so far has been minimal, with most participants in current trials staying put. This is perhaps unsurprising—since aducanumab’s approval on June 7 (Jun 2021 news), clinical uptake has been slow, held back both by confusion over how to administer the drug and lack of insurance coverage for it (see Part 1 and Part 5 of this series).
However, speakers predicted that in the future, if aducanumab becomes widely used, then trialists may have to offer participants the option of staying on it as a background therapy while also taking an investigational drug. This would greatly complicate the interpretation of trial results. Some see a silver lining, however, noting that having aducanumab will make it easier to test combination therapies, such as evaluating synergy between tau- and amyloid-based approaches.
Meanwhile, accelerated approval of the first disease-modifying therapy for AD has energized the pharma industry. Two other companies may file under the same pathway, and investors are eager to sink more money into AD research. At the same time, controversy over aducanumab’s price and irregular approval process continues to swirl in Washington, D.C., with investigations ongoing in the U.S. House and at the Department of Health and Human Services. It looks as if the earthquake of aducanumab’s approval not only shook the Alzheimer’s field, but is reverberating through U.S. healthcare and regulation more broadly (see Part 8 of this series).
So Far: Few Dropouts, Prevention Trials Plugging Along
Addressing the common concern that a drug approval prompts study participants to leave investigational drug trials for the newly available drug, Joshua Grill of the University of California, Irvine, delineated three approaches for how trials can deal with the advent of aducanumab. One, trialists could prohibit use of the drug, meaning people who want to take the antibody would have to drop out of their current trial or choose not to enroll in future trials. Two, trialists could allow aducanumab as a background therapy, similar to how acetylcholinesterase inhibitors and memantine are handled now. Three, aducanumab could be used as an active arm in the trial, perhaps replacing a placebo control. At present, most trials will take the first or second options, with the third unlikely to happen until the clinical benefit of aducanumab is established, Grill said.
How will Aduhelm being on the market affect ongoing trials? It is unethical for researchers to withhold proven therapies from trial participants. This means that for all 125 AD drugs currently in trials, researchers must notify the participants of the availability of aducanumab. To continue in their trial, participants will have to sign updated consent forms that spell out the new option. This process is underway, but has had little effect on trials so far. Grill noted there have been no dropouts at his site, and four colleagues reported either no or only one dropout at theirs.
Initially, it seemed aducanumab’s approval might prove particularly problematic for prevention trials. Because the antibody targets a presymptomatic AD pathology, people in preclinical stages of disease might be most interested in taking the drug, noted Reisa Sperling of Brigham and Women’s Hospital, Boston. Sperling runs several secondary prevention studies, including A4 and AHEAD 3-45 (Nov 2020 conference news). If many participants dropped out to take aducanumab, that could cripple a trial’s statistical power to get meaningful data. At first this seemed a real possibility, since aducanumab was approved to treat simply “Alzheimer’s disease,” meaning people with presymptomatic disease would have been eligible for treatment. After getting pushback for this, the FDA on July 8 narrowed the label to people with symptomatic AD, removing this option. “We had some sleepless nights before the label was narrowed,” Sperling quipped at AAIC.
At this time, clinicians running prevention trials do not disclose amyloid status to asymptomatic participants. That is because of the remaining uncertainty over any individual participant’s future clinical course, and because no treatments are approved for presymptomatic disease. This practice may evolve as more data emerge on an individual person’s dementia risk after a positive amyloid test, and as more treatments become available, Sperling said. For example, if a drug were to be approved to treat presymptomatic AD, then researchers would have to reveal the results of amyloid testing to participants.
What about future trials? Now that there is an approved disease-modifying therapy, are placebo-controlled trials still ethical? Yes, Grill said, because aducanumab is not yet the standard of care for AD. Not everyone will want to be on it. Once a large number of participants start dropping out to take aducanumab, that will indicate the drug has become a standard of care, he added. But he believes that time is far in the future. “It takes time to understand the implications of approved therapies. We’re not close to achieving that yet [with aducanumab],” Grill said at AAIC.
Even if aducanumab use becomes widely accepted, there will always be people who don’t qualify for the antibody, or respond poorly to it, and who want to enter trials for other drugs, Grill noted. More ethically problematic, there will also be people who want to take Aduhelm but cannot afford it. Thus, there will continue to be participants interested in placebo-controlled trials. Still, the population available for trials will likely shrink, Grill said. If people who take aducanumab differ in consistent ways from those who don’t, then those who enter trials may no longer represent the broader patient population, Sperling said.
Aducanumab As Background Therapy?
One way around these problems would be to allow trial participants to take aducanumab as a background, standard-of-care therapy. This brings with it potential problems of its own, Sperling said. For one thing, if participants in ongoing trials were allowed to start aducanumab, this change would disrupt randomization and the balance of trial arms. This problem has to be weighed against the risk of dropouts if aducanumab is disallowed. For another, researchers would need to monitor for ARIA via MRI while aducanumab was being titrated. Should dosing of the investigational drug be suspended during titration? Once participants are on aducanumab, more questions arise. Should amyloid status be monitored throughout the trial? Should aducanumab be stopped if participants become amyloid-negative? There are no answers to these questions yet, Sperling said.
A particularly thorny question that will arise in the wake of aducanumab as background drug in trials is how to interpret the trial’s data. For both clinical outcomes and adverse events, how do researchers distinguish effects due to aducanumab from those due to the investigational drug? This would be especially complicated for trials of anti-tau agents, since anti-amyloid antibodies appear to indirectly suppress tau tangles, as well. One solution might be to require all participants to take aducanumab until plaques are cleared from their brains before they start on tau therapy, Sperling suggested.
One trial coming up against this dilemma in the near future is the DIAN-TU NextGen trial, which will have three tau arms and a shared placebo group. “We are considering the impact of approved amyloid drugs,” Michael Irizarry of Eisai said at AAIC. Eisai’s anti-tau antibody E2814 is one of three investigational agents in this trial; the other two have not been selected yet (Mar 2021 news). DIAN-TU researchers are surveying participants about their interest in aducanumab and similar drugs, Irizarry said. If many want to take them, the question becomes when should an amyloid-based therapy be started—before the investigational tau drug? At the same time? After? Because DIAN-TU is a small trial, researchers need to minimize heterogeneity introduced by anti-amyloid immunotherapy treatment as best they can, Irizarry noted.
With that in mind, one proposed design is for symptomatic DIAN-TU participants to start an amyloid antibody for six months, then be randomized to a tau or placebo arm. Asymptomatic participants, on the other hand, would start in a tau or placebo arm, but be allowed to add anti-amyloid as a rescue therapy if they develop cognitive problems down the road. This design is not yet set in stone, Irizarry noted. Another option would be to use a combination design, where no anti-amyloid treatment was allowed for 12 months while testing the tau therapy, then all participants would start on it. “[Aducanumab approval] is an exciting opportunity. We hope it accelerates testing of a range of therapeutics,” Irizarry said.
What about observational studies, which track the natural course of the disease? These studies—such as ADNI, AIBL, BioFINDER, LEADS, the Mayo Clinic Study of Aging, and others—have generated invaluable data on disease progression and biomarkers. Will aducanumab and similar drugs spell the end of this type of research?
Not at all, according to David Knopman of the Mayo Clinic in Rochester, Minnesota. He believes observational studies should lean into the new landscape of AD treatment. Participants should be told they are candidates for aducanumab treatment, and allowed to start on it if they wish while staying in the study. Likewise, Knopman said, patients who have started on aducanumab should be encouraged to enter observational studies. This will provide more data on how anti-amyloid therapy changes the progression of Alzheimer’s disease.
As with prevention studies, the advent of aducanumab treatment will bring new ethical issues for observational studies. Researchers will need to tell participants their amyloid status and give them the option to go on aducanumab. Trial personnel will have to offer counseling, and participants will update their consent. Attendees at AAIC noted that this alone will be a herculean task for large studies. “The issue of reconsenting gives me shivers,” said David Morgan of Michigan State University in Grand Rapids.
What Lies Ahead for Alzheimer's Drug Discovery?
Less certain is what will happen to AD drug research in the long run. AD scientists might draw some lessons from the multiple sclerosis field, where the first drug for relapsing-remitting MS, interferon β-1b, was approved in 1993. At AAIC, Kathy Costello, Johns Hopkins Hospital in Baltimore, noted that, as with aducanumab, interferon β-1b was approved on an expedited basis and was initially controversial. The drug was expensive, at $10,000 per year, and had side effects.
Unlike aducanumab, however, interferon β-1b was recommended for people with more advanced disease. Researchers developed “exit considerations” that flagged participants in trials of other MS drugs whose symptoms became more severe, so they could leave their trials and start interferon β-1b instead. This, in turn, pushed trials toward earlier MS stages, enrolling people with milder symptoms, and led to earlier diagnosis of MS. Because aducanumab seems to work better in people at the mildest symptomatic stage, it may have the opposite effect, pushing trials of other drugs later. On the other hand, since aducanumab is currently not approved for preclinical disease, it may encourage more prevention trials, too.
More than 20 years passed before interferon β-1b replaced the placebo control arm in MS trials, in 2014. The AD field can probably expect similarly slow uptake before anti-amyloid therapy becomes the standard of care, Grill said. At that point, trials would use different designs that determine if new drugs are equivalent or superior to anti-amyloid immunotherapy. He noted that these types of trials are more complicated to run.
Sperling believes aducanumab’s approval may have some positive effects. Its attendant media attention may boost awareness of Alzheimer’s trials among the general population, aiding early diagnoses and recruitment. The availability of aducanumab will finally empower combination trials, a longtime goal of many AD researchers who believe it will take a drug cocktail to robustly fend off clinical decline (Feb 2013 conference news; Apr 2013 conference news; Mar 2014 conference news).
Specifically, researchers will be able to test the effects of combining anti-amyloid and anti-tau drugs. The Alzheimer’s Clinical Trials Consortium, which Sperling co-leads, will take action on this (Dec 2017 news). The ACTC is currently planning a trial of two anti-tau drugs. It will have six arms: anti-amyloid, tau drug 1, tau drug 2, amyloid plus tau1, amyloid plus tau2, and placebo. The plan is to run this trial placebo-controlled and double-blind for two years, then continue in open-label extension.
Participants must be at preclinical or early clinical stages and have brain amyloid and tau deposition within a defined range. The goal is to catch people at the phase of “taucatastrophe,” when tangle spread takes off beyond the confines of the entorhinal cortex and cognition starts to slide, Sperling said. The trial will investigate potential drug synergy between amyloid and tau approaches. An AAIC attendee noted the possibility that if plaques have to be cleared before tau drugs work, then simultaneous treatment might not benefit patients.
Many researchers expressed concern that aducanumab’s approval will stifle research into other types of drugs and mechanisms. That is not what happened in MS, Costello noted. Instead, interferon β-1b’s approval sparked more research into multiple disease pathways, and today, there are nearly two dozen MS drugs representing nine different mechanisms, Costello said.
Grill hopes the AD field will undergo a similar expansion. “[Aducanumab approval] is a milestone, but not the end of the journey,” he said at AAIC.
Investors are Keen, But Their Focus Remains on Amyloid
So far, aducanumab’s approval seems to have encouraged anti-amyloid approaches the most. Eli Lilly later this year will file for accelerated approval of its anti-amyloid antibody donanemab, Roche is considering doing the same for its candidate gantenerumab, and Biogen/Eisai are pushing for accelerated approval of a second antibody, lecanemab.
Those developments were no surprise. Less expected, perhaps, was AbbVie’s announcement that it, too, will advance a drug that targets plaque. “It’s quite clear that if you can remove plaque rapidly, then there will be a benefit,” company president Michael Severino said in the industry press (Endpoints News). At the same time, AbbVie is stopping its anti-tau antibody tilavonemab. This does not presage companies’ abandoning tau targets for amyloid, though, as tilavonemab had failed in progressive supranuclear palsy and targets tau’s N-terminus, which scientists now believe is ineffective (Mar 2021 news).
Meanwhile, Acumen Pharmaceuticals, whose lead candidate targets Aβ oligomers and had been slow off the starting block, scored a successful public offering, a sign that investors are eager to pour funds into anti-amyloid approaches (Endpoints News).
For their part, industry analysts are divided on whether the aducanumab approval will spur research more broadly. Some believe biomarker-based approvals will speed trials, energize researchers, and offer a roadmap for developing treatments for rare diseases (STAT news; STAT news). Others take the opposite view. Pointing out that some accelerated approvals, for example Sarepta’s Exondys 51 for muscular dystrophy, have not triggered innovation, they argue that innovation only happens when an approval deepens knowledge about disease mechanisms and encourages further research (STAT news).—Madolyn Bowman Rogers
Ahh, aducanumab ... While clinicians wrestle with administering the drug, finding out if it works, whether insurance will cover it, and how it will affect other trials (Part 1, Part 4, Part 5, and Part 7 of this series), the regulatory and U.S. public policy worlds are experiencing their own aftershocks from the FDA’s June 7 decision. The agency’s irregular process, in which it granted an accelerated approval based on biomarker data, has sparked investigations both in the U.S. House and at the Department of Health and Human Services. The latter probe will review the use of accelerated approvals more broadly, as other approvals have also come under scrutiny of late. The consequences may be felt far beyond the Alzheimer’s field.
The FDA has drawn fire from multiple sides for giving Aduhelm the nod despite inadequate efficacy data and a unanimous “no” vote from its Advisory Committee (Jun 2021 news). In the face of opposition from AdComs and its own statisticians, the agency pivoted from having considered full approval with the AdComs to granting accelerated approval based on plaque removal. On July 28 in the New England Journal of Medicine, seven AdComs members wrote that the committee was never told accelerated approval was on the table, nor asked to weigh in on that possibility. The authors, led by Caleb Alexander at Johns Hopkins Bloomberg School of Public Health, Baltimore, maintain that evidence was lacking for plaque removal as a surrogate endpoint for clinical benefit, and note that the FDA has not produced a solid scientific rationale for taking this position. “An effective surrogate should strongly correlate with a clinical endpoint in clinical studies, but an FDA statistical review … found no evidence that amyloid changes correlated with cognitive or functional changes,” they wrote. The authors took the agency to task for not seeking input on this question from the committee.
In addition, Alexander and colleagues called out that only a small number of accelerated approvals in other disease indications have subsequently produced the required evidence of clinical efficacy in post-market studies. In the case of Aduhelm, they noted that the overly broad FDA label, which does not require doctors to verify the presence of amyloid plaques, could result in Medicare wasting billions of dollars on unnecessary treatment. The authors applauded the HHS investigation as timely and needed to prevent future regulatory failures. “The overwhelming unmet need in this common and devastating disease should drive research investments, not lowering of regulatory standards that Americans rely on for safe and effective medicines,” they concluded (Alexander et al., 2021).
Scrutiny of the FDA’s process was heightened by revelations that the agency’s Billy Dunn may have had an “off-the-books,” one-on-one meeting with Biogen executive Al Sandrock at a neurology conference in May 2019, after Biogen had announced the failure of its Phase 3 trials based on an ill-advised futility analysis. As reported elsewhere, Sandrock showed Dunn the company’s updated, more-promising efficacy data, and engaged his support. After the meeting, the company established “Project Onyx,” an internal strategic effort to get the drug approved that was withheld from the agency’s advisory committee (STAT news; Endpoints News).
Other reports indicate Biogen also had a “Project Javelin,” a campaign to persuade treatment centers to administer the antibody once approved (Endpoints News).
Is all this proper? Some in Congress, such as Rep. Katie Porter (D-CA), want to find out. Porter led calls for the HHS inspector general to examine the relationship between the FDA and Biogen, and determine if rules were broken (Endpoints News). The FDA’s acting commissioner, Janet Woodcock, acknowledged the approval process had been poorly handled, and on July 9 also requested the HHS IG begin such an investigation (STAT news; Washington Post). On August 4, the IG’s office announced it would review the use of accelerated approvals more broadly, not just of aducanumab (Bloomberg; STAT news). In recent months, the accelerated approval mechanism has been criticized for greenlighting some drugs that never prove themselves, yet stay on the market (STAT news).
On June 15, Rep. Peter Welch (D-VT), wrote in an open letter to Vounatsos that “as a strong supporter of our Medicare program and an advocate for affordable as well as accessible health care, I am shocked at the price Biogen set for Aduhelm” (see Welch’s House website for the full letter).
Not to be outdone, on June 23 Sens. Elizabeth Warren (D-MA) and Bill Cassidy (R-LA) demanded their chamber’s Finance Committee hold a hearing to examine aducanumab’s price and how it will affect Medicare (full text). Thus far, the U.S. Senate has not taken up Warren and Cassidy’s call for a hearing on aducanumab. However, Senate Finance Committee Chairman Ron Wyden (D-OR) cited aducanumab while outlining principles for drug-pricing reform. “For some drugs, prices may be justified based on the remarkable clinical benefits they offer. But many, like the recently-approved Alzheimer’s drug Aduhelm, launch at prices far beyond any reasonable justification of the clinical value to patients, caregivers, or society,” Wyden wrote (Senate Committee on Finance, 2021).
All this has fueled speculation that aducanumab’s price will revive interest in drug price control legislation, and on August 12, President Biden spoke to the issue of drug prices (Washington Post; Washington Post; Washington Post).
Separately, the Aduhelm approval may damage Woodcock’s career prospects, as it has deepened Sen. Joe Manchin’s (D-WV) opposition to her appointment as FDA commissioner (Jun 2021 Manchin letter to the White House).
In response to the controversies, Dunn and drug evaluation director Patrizia Cavazzoni at the FDA have defended their decision in editorials in The Washington Post and JAMA. In the Post, they argued that such approvals have helped speed cancer research. “In a high proportion of instances, we have found that subsequent studies confirmed clinical benefit. Even though not every drug worked as expected, these approvals have propelled progress forward,” they wrote.
For its part, Biogen also pushed back on the criticism, with Sandrock penning an open letter to the AD community (Axios news). “We welcome a formal review into the interactions between the FDA and Biogen,” Sandrock wrote.—Madolyn Bowman Rogers
AL001 Boosts Progranulin. Does it Slow Frontotemporal Dementia?
Autosomal-dominant mutations underlie about 40 percent of frontotemporal dementia cases. This strong genetic component comes with a silver lining—it gives scientists a clear therapeutic target. At the Alzheimer’s Association International Conference, held July 26-30 online and in Denver, Robert Paul of Alector, South San Francisco, reported promising findings from a small open-label study testing AL001 in people with symptoms of FTD-GRN, a familial form of the disease caused by a mutation in the progranulin gene.
Paul reported that the anti-sortilin antibody restored flagging progranulin levels in the plasma and cerebrospinal fluid. Changes in other biomarkers suggested that the antibody corrected lysosomal dysfunction and soothed neuroinflammation. Over 12 months of treatment, symptoms worsened in trial participants at roughly half the rate seen in untreated volunteers in a large international cohort study, hinting at a clinical effect.
The findings offer hope that correcting progranulin deficiency halts the disease cascade, Paul said. Alector is putting AL001 to the test in an ongoing Phase 3 trial, called INFRONT3.
AL001’s target, sortilin-1, is a trafficking receptor that whisks progranulin and other proteins to the lysosome for disposal. Blocking the receptor boosts the half-life of progranulin, a protein whose concentration is more than halved in people with FTD-GRN. In the Phase 1 INFRONT study, Alector scientists found that AL001 boosted progranulin levels in the CSF of healthy volunteers and in people with FTD-GRN.
At AAIC, Paul reported 12-month findings from INFRONT-2, the open-label Phase 2 study. It treats three different cohorts: five asymptomatic GRN mutation carriers, 12 symptomatic carriers, and up to 20 symptomatic carriers of hexanucleotide expansions in the C9ORF72 gene. The latter group is still enrolling. All participants are receiving monthly infusions of 60 mg/kg AL001 for 96 weeks. The trial is small because FTD-GRN is rare, Paul said. Greater numbers of participants will be required to run the ongoing, placebo-controlled, Phase 3 trial. Only an estimated 15,000 people are known to be living with symptomatic FTD-GRN in the U.S. and Europe, the current reach of this study.
Paul presented findings from the 12 symptomatic FTD-GRN patients. First and foremost, he announced, AL001 completely rescued progranulin deficiency. Within two weeks of the first infusion, levels tripled in plasma, and remained stable out to 12 months. CSF measurements taken at baseline, six, and 12 months told a similar story, with progranulin more than doubling in response to treatment.
Dysfunctional lysosomes are a hallmark of FTD-GRN. At baseline, participants had elevated levels of two markers of lysosomal damage—cathepsin D and LAMP1—in their CSF relative to age-matched healthy controls. Both markers had dropped to control levels by six months and remained low at 12 months, Paul reported. This suggested that boosting progranulin levels had restored lysosomal function. CSF levels of the complement protein C1Q took a similar trajectory, suggesting that restoring progranulin deficiency also may have soothed damaging neuroinflammation. No other neuroinflammation markers were presented.
Did these corrections protect neurons from dying? To address this, the researchers measured fluid neurofilament light (NfL), a marker of axonal damage known to rise as FTD worsens. Here, Paul found that on average, both plasma and CSF NfL held steady throughout the trial. However, he noted that the concentration of NfL was extremely variable, both between people and within the same person from time point to time point. While there is no biological explanation yet for this variability, Paul considers the findings promising in that NfL did not appear to rise throughout the trial.
Because INFRONT-2 is an open-label study, no solid conclusions about effects on disease progression can be drawn. To attempt to put the findings in context, the researchers took advantage of data from GENFI-2, an international cohort study that tracks fluid and imaging biomarkers and clinical disease progression in people with different genetic forms of FTD (Mar 2021 conference news). Paul “randomized” 10 controls from GENFI who were matched to trial participants for age, sex, plasma NfL, and baseline scores on the CDR plus NACC-FTLD, a composite measure tailored to track FTD symptoms.
In collaboration with Howard Rosen, University of California, San Francisco, Alector scientists compared brain atrophy in AL001 trial participants and GENFI controls. As gauged by whole-brain atrophy, frontotemporal-cortical atrophy, or enlargement of the ventricles, all 22 people examined lost brain tissue over a 12-month period. That said, the people receiving AL001 lost less than did the GENFI “controls,” and the effect was most pronounced in ventricles, which expanded by half as much in AL001-treated patients as in “controls.” By contrast, some anti-amyloid treatments tested for Alzheimer’s disease, including BACE inhibitors and immunotherapies, seem to accelerate brain shrinkage (Mintun et al., 2021; Dec 2019 news).
Finally, Paul compared worsening of symptoms on the CDR plus NACC FTLD-SB. Those receiving AL001 declined 3 points, or 47 percent, less than GENFI controls over one year. Had this occurred in the context of a placebo-controlled RCT, this difference would have amounted to a clinically meaningful result, Paul claimed.
Putting the Brakes on FTD? Compared to historical controls from GENFI, trial participants receiving AL001 worsened 47 percent less on the CDR plus NACC FTLD-SB over one year. [Courtesy of Robert Paul, Alector.]
To that end, last July Alector started a Phase 3 study, called INFRONT-3. This global placebo-controlled, randomized study will test AL001 in 180 people with GRN mutations who either have symptoms or have elevated levels of plasma NfL, suggesting they are on the cusp of symptomatic disease. The trial will run for 24 months.—Jessica Shugart
While aducanumab, donanemab, and lecanemab are FDA-approved or inching closer to approval, the next generation of immunotherapies is moving through the ranks. At AAIC, scientists presented preclinical data on both passive and active immunotherapies targeting specific forms of Aβ, and even a combination vaccine against both Aβ and tau.
The latter one is a vaccine that takes aim at the top two offending proteins in AD. Robin Barbour of Prothena, South San Francisco, debuted an active dual vaccine against both proteins. Essentially, the researchers crafted three versions of the antigen used in this vaccine. Each consisted of the N-terminal 28 residues of Aβ, fused to one of three microtubule binding regions of tau. They injected mice, guinea pigs, and monkeys with the fusion proteins, along with an adjuvant. In all animals, all three constructs triggered antibody responses against both the Aβ and tau epitopes included in the vaccine. What’s more, the antibodies latched onto Aβ and tau deposits in postmortem brain tissue from people with AD.
The vaccines triggered no cytotoxic T cell responses in any of the animals—a desirable trait in such a vaccine given the potential for toxicity.
The anti-Aβ and anti-tau antibodies elicited by the vaccine showed signs of countering Aβ and tau toxicity. Antisera from immunized guinea pigs successfully blocked soluble Aβ aggregates from adhering to cultured neurons. They also blocked tau from binding heparin, considered a proxy for tau’s interaction with cell-surface heparin sulfate proteoglycans that allow tau oligomers to pass from cell to cell. Finally, antisera from mice immunized with the dual vaccine prompted cultured THP-1 cells—a human monocyte cell line—to gobble up synthetic Aβ42 protofibrils, raising the possibility that they could perhaps instigate microglia in the brain to do the same.
Another active vaccine also posted promising preclinical data at AAIC. Karen Zagorski of the Institute for Molecular Medicine, Huntington Beach, California, described a vaccine against Aβ with a pyroglutamate modification on its truncated N-terminus (pE3-Aβ). Sound familiar? Lilly’s donanemab takes direct aim at pE3-Aβ, which has only been found within amyloid plaques (Aug 2021 conference news). Zagorski cited donanemab’s success as part of the rationale behind developing an active vaccine against pE3-Aβ. Compared to a passive antibody treatment, a vaccine could theoretically prevent Aβ aggregation from happening in the first place, and would not need to be administered for months on end.
Zagorski’s pE3-Aβ vaccine was developed using Multi-TEP technology. This platform takes an antigen of choice— in this case, a series of three pE3-Aβ peptides—and attaches it to a string of antigenic peptides derived from infectious pathogens, such as tetanus and influenza. These peptides are designed to rally memory T helper cells that have seen the antigens before. Once activated, the pathogen-specific T cells crank out cytokines that give B cells a boost, improving their proliferation and production of antibodies. This platform has been used to develop vaccines against other neurodegenerative proteins, including tau and α-synuclein (Davtyan et al., 2019; Hovakimyan et al., 2019; Davtyan et al., 2017). Zagorski said that enlisting the help of memory T cells—which have recognized their cognate antigen in the past and are primed for a quick response—is particularly critical in older people, who have few remaining immunologically naïve T cells to rouse in response to a strange antigen, such as pE3-Aβ.
The Multi-TEP-P3-Aβ vaccine elicited high titers of pE3-Aβ-specific antibodies in rhesus macaques. Zagorski reported that immunized 5xFAD transgenic mice also churned out antibodies specific to pE3-Aβ, but not to unmodified Aβ42. Nevertheless, the total level of insoluble human Aβ42—including all forms of the peptide—halved in the brains of treated mice. This suggested that the pE3-Aβ-specific antibodies also instigated the clearance of unmodified forms of the peptide that co-mingle with the pyroglutamate-Aβ.
On this note of sweeping out types of Aβ that mingle together, Prothena presented preclinical data that PRX012, a fully humanized IgG1 antibody that binds Aβ at its N-terminus, clears pE3-Aβ as well. Wagner Zago and colleagues had wondered how PRX012 compared in this regard to aducanumab, which also binds the N-terminus of Aβ. They also wondered if PRX012 had cleared this toxic form of the peptide by binding to it.
To address the latter question, the researchers used an ELISA, and found that while PRX012 binds to full-length Aβ42 with high affinity, it does not bind to pE3-Aβ. Next, they used surface plasmon resonance spectroscopy to compare the binding characteristics of PRX012 to those of aducanumab. The technique gauges interactions between an immobilized ligand and its partner by detecting changes in light caused by jostling electrons on a sensor surface. They found that PRX012 bound synthetic, full-length Aβ42 fibrils with 10-fold higher avidity than did aducanumab. The scientists did not report if this aducanumab, which they produced themselves, bound pE3-Aβ, although a previous study reported that aducanumab binds to the pE3-Aβ with 31-fold lower affinity than it does to the unmodified, full-length peptide (Arndt et al., 2018).
If PRX012 does not bind pE3-Aβ, then how can it clear it from the brain? Using immunohistochemistry, the scientists found that PRX012 glommed onto Aβ plaques within tissue sections from AD brain, including onto the dense cores where pE3-Aβ abounds. When primary mouse microglia were added to the sections along with PRX-012, the cells ate up both forms of Aβ—nearly halving Aβ42 fibrils and reducing pE3-Aβ by roughly three-quarters. PRX012 instigated the microglial mop-up of pE3-Aβ at roughly ninefold lower concentration than did aducanumab. Zago presented no comparison of PRX012 to donanemab, which specifically targets pE3-Aβ. His company aims to test PRX012 in clinical trials starting in 2022.
Notably, the amount of PRX012 needed to clear pE3-Aβ from the brain tissue sections was on par with the concentration expected in the brain following a subcutaneous injection. Compared to the intravenous infusions currently used for other immunotherapies, including aducanumab, subcutaneous injections would ease the burden of treatment because they can be administered at home.—Jessica Shugart
ADAD and LOAD: At Cellular Level, They Are Not the Same
At this year’s Alzheimer’s Association International Conference, scientists reported the first substantial single-nucleus RNA-Seq analysis of brain tissue from people who had had familial Alzheimer’s disease. They found subclusters among all main cell types that were unique to autosomal-dominant forms of AD. Similarly, they found subclusters enriched in people who carried known risk variants for late-onset AD. The ADAD and LOAD subclusters were not the same, suggesting perhaps that the brain mounts different cellular responses to different forms of AD.
Also at AAIC, researchers debuted one of the largest subclustering analyses of microglia to date, based on approximately 90,000 nuclei. Previously identified disease-associated transcriptome signatures did not fall neatly into any one cluster. This implies it may be difficult to therapeutically target specific subclusters of microglia.
ADAD vs. LOAD
The ADAD snRNA-Seq data came from the lab of Bruno Benitez and Oscar Harari at Washington University, St. Louis. First author Logan Brase and colleagues isolated almost 300,000 nuclei from samples of parietal lobes taken postmortem from 67 volunteers registered at the Knight ADRC and DIAN brain banks. Sixteen of these donors had had ADAD, carrying mutations in either the amyloid precursor protein gene or presenilin 1. Other samples came from nine healthy controls, eight people with other diseases, three with presymptomatic AD, and 31 who had had sporadic AD. The latter included people who carried one of four different genetic variants of TREM2, or the rs1582763 protective single nucleotide polymorphism that lies near the MS4A locus.
The Usual Blotches. From almost 300,000 nuclei, snRNA-Seq analysis identifies the main cell types in the brain. [Image courtesy Logan Brase, WashU.]
Brase analyzed gene expression patterns to identify subclusters of brain cells. He found nine distinct types of microglia among 17,089 nuclei, five subclusters among 38,815 astrocytes, four types of excitatory neurons among 38,176, and three subtypes from 15,414 inhibitory neurons. However, looking specifically at nuclei from people who had had ADAD, the subclustering pattern was different.
Among their microglia, subcluster 4 stood out as an ADAD-specific cadre. It predominated in ADAD but was teeny in late-onset AD samples, and all but absent from healthy controls, people with presymptomatic AD, and from brains with other diseases (see image below). Brase found that 5xFAD mouse brain samples contained a microglial subcluster analogous to the human subcluster 4, hinting that the APP/PS mutations might drive the expression signature—5xFAD mice contain three APP and two PS1 mutations.
What does microglial subcluster 4 do? It upregulates genes involved in lipid biology/atherosclerosis, autophagy, and necroptosis, Brase told Alzforum, though he does not know yet what it does in ADAD. Curiously, this cluster bore little resemblance to the DAM, MGnD, and HAM microglial transcriptomes previously linked to AD (Jun 2017 news; Sep 2017 news; May 2019 news). Instead, those latter AD-related genes overlapped with transcripts upregulated in Brase’s microglial subcluster 1. This subcluster was not enriched in ADAD, but was well-represented in all brain samples analyzed.
Not only microglia distinguished themselves in ADAD. Three oligodendrocyte precursor cell subclusters, two astroglial subclusters, and one each of the oligodendrocyte and neuronal subclusters were enriched in samples from FAD mutation carriers. The transcriptome of one of the astrocyte subclusters resembled disease-associated astrocytes that were recently identified in transgenic mouse models of AD and in human brains (Habib et al., 2020).
Enriched in ADAD. Among nine microglial subclusters found in ADAD (left), cells in cluster 4 were the most plentiful (orange scale, see asterisk). This cluster was barely detectable (right) in samples from controls (CO), sporadic AD (sAD), presymptomatic AD (Pres), or people with other diseases (OTH). (Asterisk denotes significant differences.) [Image courtesy Logan Brase, WashU.]
What about sporadic AD? The scientists found no subclusters enriched in this group as a whole. However, when they looked at nuclei from people who carried genetic risk variants, differences did pop out. Brains from people with either the R47H, R62H, or H157Y mutations that reduce TREM2 activation had distinct microglial and oligodendrocyte profiles. Specifically, they contained more microglial subcluster 2 and oligodendrocyte subcluster 5 cells. The researchers saw the same pattern among TREM2 carriers in the Religious Orders Study and Memory and Aging Project (ROSMAP) data set.
For the most part, what biological effect these cellular shifts might have in AD remains to be seen. The oligodendrocyte subcluster 5 cells increased expression of TFEB, a regulator of the autophagy/lysosomal pathway that can become compromised in many neurodegenerative disorders. Other WashU scientists have found that these TREM2 variants tone down gene expression responses within microglial clusters in samples from AD prefrontal cortices (Jan 2020 news).
In contrast to the R47H/R62H/H157Y cases, one person who carried the R136W TREM2 mutation had subcluster densities for microglia, astrocytes, oligodendrocyte precursors and oligodendrocytes that more closely matched the profile of ADAD. “It’s hard to draw any definitive conclusions about this rare mutation because we only had a single subject,” Benitez told Alzforum. “We don’t know what that mutation does, but it would be really interesting to see if it might have a gain of function.” This person was diagnosed with early onset AD, i.e., before age 60, so there could be some links between ADAD to explore, said Benitez.
Next, Brase and colleagues studied the rs1582763 SNP at the MS4A locus. An adenine (A) for a guanine (G) nucleotide swap protects against AD. Here again, carriers shared a unique microglial profile. The GG homozygotes had no microglia subcluster 3. Only a few microglia sorted into this subcluster in the AG heterozygotes, while the cluster bloomed eightfold in the AA homozygotes. The same pattern repeated in the ROSMAP sample, albeit weakly.
Subcluster 3 microglia exhibited a transcription profile consistent with activation, upregulating genes such as NPC1 and TGFβ receptors. Curiously, subcluster 3 comprised nearly half of all microglia in people who had been diagnosed with presymptomatic AD, making these cells fivefold more abundant than in controls. It had all but vanished in LOAD and was absent in ADAD, hinting that perhaps this subcluster reflects an attempt by microglia to combat pathology early in the disease process.
Finally, Brase leveraged this subcluster dataset to test if expression of other LOAD variants might be important to specific cell types. While geneticists have identified at least 75 genetic variants that associate with AD in genome-wide analysis, they do not know what the functional locus is for most of them, or what cell types express the disease-associated variant (Feb 2021 news).
Brase focused on expression of several dozen of the most notable GWAS hits, including Bin1, SORL1, PICALM, etc., in cell subclusters. “The idea is that if a gene is differentially expressed in subcluster 1 vs. subcluster 2, then that would highlight the importance of that gene in that cell type,” he said. In this analysis, some expression patterns turned out as expected. For example, differential expression of APOE was high among astrocytes and microglia, cells known to produce ApoE.
But there were surprises, too. PLGC2, thought to be predominantly microglial, was differentially expressed across the board, and strongly in neurons and oligodendrocytes. SORL1, typically associated with neurons, had a similar pattern. “The findings suggest that many GWAS genes may have a much broader role in disease than in the cells we typically associate them with,” said Brase.
It's Not All About Subclusters
Cell subclusters are defined by their shared preponderance of commonly expressed genes, but that doesn’t mean the expression pattern within a given subcluster is fixed. Katherine Prater, who works in the lab of Suman Jayadev at the University of Washington, Seattle, emphasized this when she presented one of the largest subclustering analyses of microglia to date.
Prater and colleagues got around a central problem facing scientists who study microglia using snRNA-Seq, namely that these cells represent only 3 to 5 percent of the nuclei typically recovered from the human brain. Hence, some of the early RNA-Seq analysis of AD microglia came from as few as several hundred or thousand cells. To grow that paltry catch, Prater and colleagues developed a fluorescent activated sorting method that homes in on nuclei carrying the essential microglial transcription factor PU.1.
Their system worked. Prater greatly boosted the number of microglia isolated from brain. In samples from four people, she was able to identify about 1,000 microglial nuclei using traditional methods and 23,300 after PU.1 sorting.
Prater used this enrichment method to isolate microglial nuclei from flash-frozen prefrontal cortices obtained postmortem from 22 people, average age 86. Twelve had had AD as determined by neuropathology, 10 were controls. Prater isolated about 4,000 microglia nuclei from each sample for a whopping 90,000 in toto. Researchers in Denmark recently achieved a similar enrichment, not by capturing microglial nuclei, but by throwing away those that come from neurons or oligodendrocytes (Gerrits et al., 2021).
Prater identified 10 microglial subclusters, each with distinct expression profiles that suggest specific functions, such as homeostasis, cell cycling, stress responses, immune responses, and cell death. Notably, some subclusters that had been identified from previous small hauls of microglia did not emerge here (May 2019 news; Jun 2017 news). Instead, their expression signatures spread across several of Prater’s subclusters. DAMs fit this scenario. “We probably don’t want to be fixated on DAMs,” Prater told Alzforum. “There is more to the changes seen in AD than just ‘DAMs appear,’ or ‘DAMS are dysregulated.’ Other subtypes are changing that we can’t just ignore.”
Indeed, Prater found that even within a given subcluster, differences emerge in AD. Cells in the homeostatic cluster, for example, were more likely to express antigen-processing and antigen-presenting genes, or immune response or endocytosis genes if the cells came from a donor with AD. Ditto for cells in the metabolism and endocytosis microglia subcluster.
“We see in AD an enrichment of inflammatory pathways and genes across many, but not all subtypes of microglia,” Prater said. “We need a lot more data to find out if we can target specific subtypes, or if it’s even possible to influence ‘bad’ microglia without affecting other microglia.”—Tom Fagan
Polygenic Scores Paint Microglia as Culprits in Alzheimer's
Mere responders no more, microglia nowadays are respected—if not feared—as early actors when a person is developing a neurodegenerative disease. Several presentations at last month’s Alzheimer’s Association International Conference underscored their pre-eminent role within the pathogenetic cascade of Alzheimer’s disease. Using a cell-type-specific version of a polygenic risk score, researchers reported that myriad genetic variations that are active in microglia can exacerbate amyloid deposition. In contrast, AD-related polygenic variation that is active in neurons has more impact on cognition. To chart how genetic variation alters the expression of microglial genes and influences AD and related diseases, scientists introduced the microglial genomic atlas (MiGA), the largest repository of human microglial transcriptomes to date. The atlas helped nail down associations between AD risk variants and microglial genes, which previously had been inferred from studies of other myeloid cell types.
Massive genome-wide association studies have uncovered dozens of genetic polymorphisms that influence the risk of Alzheimer’s disease (Feb 2021 news). Most of these individual variants impart only a smidgen of risk, but collectively, they exert a strong influence. Polygenic risk scores are calculated by tallying the effects of multiple risk variants. They have been tied to all manner of AD-related phenotypes, including plaques, tangles, brain atrophy, and cognitive decline (Escott-Price et al., 2015; Jul 2016 news; Feb 2019 news).
Lost in this polygenic fray is the question of exactly which cells these cumulative risk variants influence. After all, most, if not all, AD risk variants regulate expression in specific cell types. This was the main theme of Hyun-Sik Yang, Brigham and Women’s Hospital in Boston. Wondering how genetic variants active in different cell types contribute to different aspects of preclinical AD, Yang accessed baseline data from the A4 study, a secondary prevention trial that is testing solanezumab in people with brain amyloid who were cognitively normal at baseline. All 2,961 A4 participants had consented to genome-wide genotyping.
Yang and colleagues started by calculating an overall polygenic risk score for each participant based on the variants he or she carried. Using risk variants identified in the largest AD GWAS to date, the researchers employed a Bayesian statistical method that accounts for individual variations in genomic architecture to calculate each person’s PRS (Ge et al., 2019). The analysis incorporated contributions from thousands of genetic variants, most of which fell below a stringent threshold for genome-wide significance.
Crucially, for each variant the scientists referenced single-cell RNA sequencing data from postmortem brain samples in order to designate which cell types expressed the putative causal gene (Mathys et al., 2019). This enabled them to derive distinct polygenic risk scores for each of eight different cell types: excitatory neurons, inhibitory neurons, oligodendrocytes, oligodendrocyte progenitors, astrocytes, microglia, endothelial cells, and pericytes. Some genetic variants were active in more than one cell type. Ultimately, each A4 participant was assigned nine different PRSs: an overall one that included all variants, and one for each of the eight cell types.
Which PRS had the strongest ties to baseline amyloid levels among all the A4 participants? The microglial score was the clear winner in this regard, Yang said. Its association with baseline Aβ load was about twice as strong as either neuronal PRS, and topped the overall PRS by a third. This finding cast microglia as intimately involved in amyloid.
Microglia Drive Plaque. Among people in the A4 study, their microglia-specific PRS scores had the strongest association (y axis) with baseline amyloid burden. [Courtesy of Hyun-Sik Yang, Brigham and Women’s Hospital.]
The story was different for cognition. Using participants’ baseline scores on the preclinical AD cognitive composite (PACC) as a gauge, Yang found that more than any other PRS, a person’s excitatory neuron PRS was most strongly tied to his or her cognitive performance at study start. The higher a person’s excitatory neuron PRS, i.e., the more risk variants known to affect genes in these neurons, the lower he or she tended to perform on the test. The strength of this relationship dropped only slightly when adjusted for Aβ burden, suggesting that cumulative AD risk variants active in excitatory neurons influence cognition independently of amyloid. Yang thinks these variants could influence AD risk by weakening neuronal function. Akin to the concept of cognitive reserve, a high neuronal PRS could render neurons more susceptible to a given amount of amyloid.
Julie Williams of Cardiff University in Wales commented that Yang’s findings convincingly show that microglia are part of the causative pathway of AD, especially since A4 participants were at the earliest stages of the disease.
Lianne Reus of Amsterdam University Medical Center also took a polygenic approach to investigate how different cell types influence AD pathogenesis. At AAIC, Reus linked cell-type-specific PRS to fluid biomarkers. She drew genotyping data from more than 617 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and 918 from the European Medical Information Framework for Alzheimer’s Disease (EMIF-AD) for a total of 525 controls, 709 people with MCI, and 272 with AD. (Shaw et al., 2009; Bos et al., 2018).
To calculate cell-type-specific PRSs for each person, Reus took a more candidate-based approach. Instead of weaving thousands of variants into her analysis, Reus selected only 26. These had risen to genome-wide significance in GWAS, and had detectable levels of RNA in at least one brain-cell type in the BRAIN-RNA-Seq database (de Rojas et al., 2021; Zhang et al., 2014). Reus designated a gene as cell-type specific if at least 50 percent of its expression came from a single cell type. Of the 26 variants, 10 were nonspecific, 11 were microglia-specific, four were astrocyte-specific, one was specific to neurons, and none were specific for endothelial cells or oligodendrocytes (see genes in image below). With this minimalist panel, Reus calculated four PRSs for each person: one for the 10 nonspecific loci, and one each for microglia, astrocytes, and even neurons, though this was based on only one variant. She then asked how each person’s four PRSs associated with their CSF concentration of Aβ, total tau, p-tau181, neurogranin, neurofilament light (NfL), and YKL40.
Getting Specific. Of 26 AD risk variants (right), 10 were expressed across cell types, 11 were more expressed in microglia, four in astrocytes, and one was found expressed only in neurons. [Courtesy of Lianne Reus, Amsterdam University.]
ApoE was designated as an astrocyte-specific gene. When included in the PRS for that cell type, the astrocyte polygenic scores strongly associated with CSF Aβ, total tau, p-tau181, and neurogranin. Without ApoE, a person’s astrocyte PRS, based on only three possible variants, associated only with CSF Aβ, demonstrating the power of ApoE’s connection to AD pathogenesis more broadly. The microglial PRS also associated with CSF Aβ, and nominally with total tau. Neither the neuron-specific score nor the nonspecific PRS was significantly tied to any CSF biomarker. Notably, only one variant in SLC24A4 was part of the neuronal “PRS” in this dataset.
Although Yang and Reus’s studies employed different methods to calculate PRS and to designate cell-type specificity of AD risk variants, they drew one common conclusion, namely that markers of amyloid deposition associate with variants expressed in astrocytes and/or microglia.
Glial Ties to Aβ. The astrocyte PRS linked to several CSF biomarkers (right) with high statistical significance. Without ApoE, the astrocyte PRS only linked to CSF Aβ. Microglial PRS associated with CSF Aβ and nominally with total tau. [Courtesy of Lianne Reus, Amsterdam University.]
“Cell-type-specific polygenic risk scores help to clarify the contribution of specific cell types in AD pathophysiology in different stages of the disease,” wrote Betty Tijms of Amsterdam University Medical Center and Pieter-Jelle Visser of Maastricht University in the Netherlands, who co-authored the study with Reus. “The precise cell-type-dependent interactions will probably vary between individuals, and so this approach may help to better understand interindividual variation in disease manifestation.”
Microglia Atlas Connects the Dots
While the central role of microglial variants in AD has become clear, exactly how most of them influence microglial gene expression and cellular function is unclear. That is partly because microglia are so changeable. They adapt quickly to changes in their environment. Genetic variation could therefore affect microglia differently depending on which brain region they are in, their age, or their response to a pathological condition. What’s more, because genetic variants rarely occur within the coding region of a gene, and gene expression varies dramatically by cell type, detailed transcriptomic data are needed to nail down microglia-specific relationships between variants and gene expression. This requires large numbers of human microglial samples. These are in short supply, hence researchers have used transcriptomic data from other myeloid cells, or they have made do with live microglia extracted from small numbers of people during brain surgery to chart connections between genetic variants and gene expression in the cells (Aug 2019 news; Jun 2017 news; Nov 2019 news).
Mega MiGA? Transcriptomes of 255 live microglial samples, taken from 100 human donors at autopsy, form expression profiles from four brain regions. MiGA could serve a number of purposes, including prioritization of disease genes. [Courtesy of Lopes et al., bioRxiv, 2021.]
At AAIC, Katia Lopes of the Icahn School of Medicine in New York presented initial findings of transcriptomes garnered from one of the larger brain samples to date. Called the microglia genomic atlas (MiGA), the trove was built using 255 tissue samples taken from 100 human donors at autopsy. The donors included people with no signs of neurological disease, as well as those with AD, PD, and other neurological disorders. Live microglia were teased from brain samples collected from four brain regions: the medial frontal gyrus and superior temporal gyrus in the cortex, and the thalamus and subventricular zone. Researchers led by Robin Franklin at the University of Cambridge and Daniel Gaffney at the Wellcome Sanger Institute, Hinxton, both in the U.K., recently profiled transcriptomes of live microglial among 112 tissue samples acquired from 141 donors undergoing neurosurgery (Young et al., 2021).
Sex did not significantly alter microglial profiles, but age did: The researchers detected nearly 1,700 genes whose expression in these cells changes with age. Genes upregulated in aging included those involved in lipid metabolism and immune responses, while those that flagged with age play a role in cell motility, polarity, and IL-6 cytokine signaling. Intriguingly, some of the genes that lessen with age overlapped with those reported to dampen microglia expression in people who have AD (Srinivasan et al., 2020).
At AAIC, Lopes showed results from a quantitative trait loci (QTL) analysis. Essentially, the scientists looked for genetic polymorphisms that influenced either the expression or splicing of genes in microglia. The eQTLs modulate expression of genes that are dubbed eGenes; sQTLs influence splicing of sGenes. By combining microglial transcriptome data from all four brain regions sampled, the researchers identified 3,611 eGenes and 4,614 sGenes. Most of these were expressed across brain regions, but some were specific. For example, expression of the RNF40 gene was influenced by one eQTL, but only in microglia inhabiting the subventricular zone.
Did any of the eQTLs or sQTLs identified in MiGA influence genes associated with AD risk loci? Indeed, the researchers found 15 MiGA QTLs that influenced expression and/or splicing of genes associated with 10 AD risk loci, meaning that some loci had more than one QTL. When more than one gene associates with a given risk locus, researchers cannot always tell which gene is driving the risk. Lopes and colleagues leveraged the co-localization of QTLs with risk loci to help answer this question.
Indeed, for one such AD risk locus—ECHDC3—Lopes used data from MiGA to zero in on the gene influenced by the variation. The lead AD risk SNP resides between two genes in the locus: ECHDC3 and USP6NL. In MiGA, Lopes identified an eQTL within the locus that associated with increased expression of USP6NL, and was also nominally tied to increased AD risk.
Using fine mapping of the locus, she found three additional SNPs associated with higher AD risk. Considering the AD risk of SNPs and eQTL SNP combined, four of the five landed within a microglial enhancer. Finally, using published microglial proximity ligation assisted ChIP-Seq (PLAC-Seq) data, which essentially catches an enhancer in the act of hooking up with a gene, Lopes found that the enhancer associated with USP6NL, but not ECHDC3 (Nott et al., 2019).
In short, three different forms of data converged on USP6NL as the causal gene. “Now that we have all this information—eQTL, co-localization with risk SNPs, and PLAC-Seq—it is exciting to overlay all of these to find the mechanism behind the altered expression,” Lopes said.
MiGA demonstrated that many neurological disease susceptibility loci are mediated through gene expression or splicing in microglia, and suggested causal variants, noted Valentina Escott-Price of Cardiff University. The atlas may therefore help further refine gene and SNP lists to make the polygenic risk scores—which rely on estimations of causal genes—more relevant to the disease and improve prediction for eventual use in people.—Jessica Shugart
Bos I, Vos S, Vandenberghe R, Scheltens P, Engelborghs S, Frisoni G, Molinuevo JL, Wallin A, Lleó A, Popp J, Martinez-Lage P, Baird A, Dobson R, Legido-Quigley C, Sleegers K, Van Broeckhoven C, Bertram L, Ten Kate M, Barkhof F, Zetterberg H, Lovestone S, Streffer J, Visser PJ.
The EMIF-AD Multimodal Biomarker Discovery study: design, methods and cohort characteristics.
Alzheimers Res Ther. 2018 Jul 6;10(1):64.
PubMed.
de Rojas I, Moreno-Grau S, Tesi N, Grenier-Boley B, Andrade V, Jansen IE, Pedersen NL, Stringa N, Zettergren A, Hernández I, Montrreal L, Antúnez C, Antonell A, Tankard RM, Bis JC, Sims R, Bellenguez C, Quintela I, González-Perez A, Calero M, Franco-Macías E, Macías J, Blesa R, Cervera-Carles L, Menéndez-González M, Frank-García A, Royo JL, Moreno F, Huerto Vilas R, Baquero M, Diez-Fairen M, Lage C, García-Madrona S, García-González P, Alarcón-Martín E, Valero S, Sotolongo-Grau O, Ullgren A, Naj AC, Lemstra AW, Benaque A, Pérez-Cordón A, Benussi A, Rábano A, Padovani A, Squassina A, de Mendonça A, Arias Pastor A, Kok AA, Meggy A, Pastor AB, Espinosa A, Corma-Gómez A, Martín Montes A, Sanabria Á, DeStefano AL, Schneider A, Haapasalo A, Kinhult Ståhlbom A, Tybjærg-Hansen A, Hartmann AM, Spottke A, Corbatón-Anchuelo A, Rongve A, Borroni B, Arosio B, Nacmias B, Nordestgaard BG, Kunkle BW, Charbonnier C, Abdelnour C, Masullo C, Martínez Rodríguez C, Muñoz-Fernandez C, Dufouil C, Graff C, Ferreira CB, Chillotti C, Reynolds CA, Fenoglio C, Van Broeckhoven C, Clark C, Pisanu C, Satizabal CL, Holmes C, Buiza-Rueda D, Aarsland D, Rujescu D, Alcolea D, Galimberti D, Wallon D, Seripa D, Grünblatt E, Dardiotis E, Düzel E, Scarpini E, Conti E, Rubino E, Gelpi E, Rodriguez-Rodriguez E, Duron E, Boerwinkle E, Ferri E, Tagliavini F, Küçükali F, Pasquier F, Sanchez-Garcia F, Mangialasche F, Jessen F, Nicolas G, Selbæk G, Ortega G, Chêne G, Hadjigeorgiou G, Rossi G, Spalletta G, Giaccone G, Grande G, Binetti G, Papenberg G, Hampel H, Bailly H, Zetterberg H, Soininen H, Karlsson IK, Alvarez I, Appollonio I, Giegling I, Skoog I, Saltvedt I, Rainero I, Rosas Allende I, Hort J, Diehl-Schmid J, Van Dongen J, Vidal JS, Lehtisalo J, Wiltfang J, Thomassen JQ, Kornhuber J, Haines JL, Vogelgsang J, Pineda JA, Fortea J, Popp J, Deckert J, Buerger K, Morgan K, Fließbach K, Sleegers K, Molina-Porcel L, Kilander L, Weinhold L, Farrer LA, Wang LS, Kleineidam L, Farotti L, Parnetti L, Tremolizzo L, Hausner L, Benussi L, Froelich L, Ikram MA, Deniz-Naranjo MC, Tsolaki M, Rosende-Roca M, Löwenmark M, Hulsman M, Spallazzi M, Pericak-Vance MA, Esiri M, Bernal Sánchez-Arjona M, Dalmasso MC, Martínez-Larrad MT, Arcaro M, Nöthen MM, Fernández-Fuertes M, Dichgans M, Ingelsson M, Herrmann MJ, Scherer M, Vyhnalek M, Kosmidis MH, Yannakoulia M, Schmid M, Ewers M, Heneka MT, Wagner M, Scamosci M, Kivipelto M, Hiltunen M, Zulaica M, Alegret M, Fornage M, Roberto N, van Schoor NM, Seidu NM, Banaj N, Armstrong NJ, Scarmeas N, Scherbaum N, Goldhardt O, Hanon O, Peters O, Skrobot OA, Quenez O, Lerch O, Bossù P, Caffarra P, Dionigi Rossi P, Sakka P, Hoffmann P, Holmans PA, Fischer P, Riederer P, Yang Q, Marshall R, Kalaria RN, Mayeux R, Vandenberghe R, Cecchetti R, Ghidoni R, Frikke-Schmidt R, Sorbi S, Hägg S, Engelborghs S, Helisalmi S, Botne Sando S, Kern S, Archetti S, Boschi S, Fostinelli S, Gil S, Mendoza S, Mead S, Ciccone S, Djurovic S, Heilmann-Heimbach S, Riedel-Heller S, Kuulasmaa T, Del Ser T, Lebouvier T, Polak T, Ngandu T, Grimmer T, Bessi V, Escott-Price V, Giedraitis V, Deramecourt V, Maier W, Jian X, Pijnenburg YA, EADB contributors, GR@ACE study group, DEGESCO consortium, IGAP (ADGC, CHARGE, EADI, GERAD), PGC-ALZ consortia, Kehoe PG, Garcia-Ribas G, Sánchez-Juan P, Pastor P, Pérez-Tur J, Piñol-Ripoll G, Lopez de Munain A, García-Alberca JM, Bullido MJ, Álvarez V, Lleó A, Real LM, Mir P, Medina M, Scheltens P, Holstege H, Marquié M, Sáez ME, Carracedo Á, Amouyel P, Schellenberg GD, Williams J, Seshadri S, van Duijn CM, Mather KA, Sánchez-Valle R, Serrano-Ríos M, Orellana A, Tárraga L, Blennow K, Huisman M, Andreassen OA, Posthuma D, Clarimón J, Boada M, van der Flier WM, Ramirez A, Lambert JC, van der Lee SJ, Ruiz A.
Common variants in Alzheimer's disease and risk stratification by polygenic risk scores.
Nat Commun. 2021 Jun 7;12(1):3417.
PubMed.
Correction.
Young AM, Kumasaka N, Calvert F, Hammond TR, Knights A, Panousis N, Park JS, Schwartzentruber J, Liu J, Kundu K, Segel M, Murphy NA, McMurran CE, Bulstrode H, Correia J, Budohoski KP, Joannides A, Guilfoyle MR, Trivedi R, Kirollos R, Morris R, Garnett MR, Timofeev I, Jalloh I, Holland K, Mannion R, Mair R, Watts C, Price SJ, Kirkpatrick PJ, Santarius T, Mountjoy E, Ghoussaini M, Soranzo N, Bayraktar OA, Stevens B, Hutchinson PJ, Franklin RJ, Gaffney DJ.
A map of transcriptional heterogeneity and regulatory variation in human microglia.
Nat Genet. 2021 Jun;53(6):861-868. Epub 2021 Jun 3
PubMed.
Mirror, Mirror on the Wall, Who’s the Earliest of Them All?
At last month's Alzheimer’s Association International Conference, scientists showed new data that confirms phospho-tau231 as one of the earliest biomarkers known to rise in the plasma of people with preclinical Alzheimer’s disease, but is it really the first? According to new data also presented there, glial fibrillary acidic protein and YKL-40, two markers of astrogliosis, may rise even sooner. The findings imply that neuroinflammation begins decades before symptoms, and that astroglial markers could help identify people at risk and track their response to therapies.
“The take-home message was that you can track ‘mediators’ of Aβ toxicity using astrocytic and microglial biomarkers,” said Henrik Zetterberg, University of Gothenburg, Sweden. “This should be helpful in therapeutic trials not focusing on Aβ.”
Alas, GFAP tells a confusing story. At AAIC, scientists confirmed that levels in the plasma, but not in the CSF, correlate with amyloid plaque load, leaving them searching for an explanation.
GFAP and YKL-40 are predominantly markers of astrogliosis. Scientists have long known they tick up in AD but exactly when that starts is not clear. Yi Ting (Tina) Wang, working in the lab of Pedro Rosa-Neto at McGill University, Montreal, wanted to chart how neuroinflammatory markers behave across the AD continuum, and how they compare to markers of amyloid, tau, and neurodegeneration. Wang used PET to measure binding of the amyloid ligand AZD4694 in 210 volunteers in the TRIAD primary care cohort established by Rosa-Neto. Then, she used PET standard uptake value ratios (SUVRs) for each person to approximately pinpoint how far along they are in the disease continuum. Then using the SUVRs as a proxy for progression, she charted when GFAP, YKL-40, and other markers in the CSF begin to change. Hers was a cross-sectional analysis. “The proxy analysis gives you an idea of what the progression might look like, but we need longitudinal data to confirm,” Wang told Alzforum.
What did this first look reveal? For one, Wang noticed that CSF GFAP and YKL-40 were high, low, then high again in what appeared to be a biphasic fashion in people on different points along the progression to AD dementia. She believes the first wave indicates a stage where cells are responding to the beginning of amyloidosis; the second, Wang thinks, may reflect ineffective clearance of plaques followed by the emergence of tau pathology. The first wave would be in keeping with astrocytosis discovered very early in AD by PET (Feb 2012 news).
How do these waves resonate with other markers? Setting SUVR of 1.5 as a cutoff for amyloid positivity, Wang found that GFAP and YKL-40 began to rise very early in people who were amyloid-negative (see image below). In fact, the increase in GFAP and YKL-40 coincided with change in the CSF Aβ42/40 ratio, which falls before PET scans turn positive. Phospho-tau231 rose steeply soon after that, as did, to a lesser extent, p-tau181 and p-tau217. The order fits with data published earlier this year by Rosa-Neto and Zetterberg’s group, but see below on p-tau231 findings (Feb 2021 news).
Making Waves. Compared to amyloid PET SUVR, a proxy for disease progression, GFAP and YKL 40 rise very early in the AD continuum, fall, then rise again as plaques (top) and tangles (bottom) accumulate. The neuroinflammation markers rise earlier and reach higher concentrations in the CSF than all other markers bar NfL. [Courtesy of Tina Wang, McGill University.]
Correlating CSF and imaging markers pair-wise with each other suggested that astrocytes may indeed be reacting to waves of amyloid and tangles. In 95 volunteers without plaques or tangles, i.e., who were A-/T- in the ATN classification of amyloid, tau, and neurodegeneration markers, CSF GFAP and YKL40 did not correlate with Aβ42 and only moderately correlated with Aβ40 and p-tau181. In 23 A+/T- volunteers, i.e., people with plaques but no tangles, the inflammation markers moderately to strongly correlated with Aβ42 and with total tau. And in 62 A+/T+ participants, CSF YKL40, but not CSF GFAP, correlated with tau PET and p-tau181.
Two waves mean that trying to use these inflammatory markers in individual patients will require great care. “We need to know how to interpret the data by looking at correlations with Aβ and tau, because neuroinflammation can be caused by many other conditions,” said Wang.
Alternatively, scientists may just turn to plasma GFAP. In her talk, Wang did not discuss how this marker behaves, but her abstract does not show the same biphasic pattern (see image at left).
Others have seen the same puzzling divergence, namely that plasma GFAP correlates more tightly with AD pathology than does CSF GFAP. At ADPD last March, Andrea Benedet, in the labs of Zetterberg and Kaj Blennow at UGothenburg, reported this for 171 volunteers in the TRIAD cohort (Mar 2021 news). The revelation that a plasma solute might outperform its alter ego in the CSF took the field by surprise; after all, doesn't the former drain from the latter? Indeed, CSF markers have previously shown stronger correlations with brain pathology than their plasma counterparts.
Benedet and colleagues, working with Marc Suárez-Calvet at the Barcelonaβeta Brain Research Center in Spain have since confirmed their TRIAD findings in the ALFA+ study cohort in Spain and the BioCogBank Paris Lariboisière cohort in France. Their paper is in press.
Meanwhile at AAIC, Joana Pereira, from Stockholm's Karolinska Institutet, confirmed that plasma outdid CSF in evaluating GFAP 504 volunteers in the Swedish BIOFINDER cohort. Collaborating with Oskar Hansson’s group at Lund University, Pereira found more GFAP in plasma of people who tested positive for amyloid as per their CSF Aβ42/40 ratio, than in those who were negative, regardless of cognitive status. The rise in plasma GFAP seemed tied to Aβ pathology because plasma GFAP was normal even among 75 volunteers diagnosed with a non-AD dementia, such as frontotemporal, dementia with Lewy bodies, Parkinson’s, and progressive supranuclear palsy. CSF GFAP, on the other hand, rose in this non-AD group, but was unable to distinguish amyloid-positive from amyloid-negative people (see image below).
“We’ve had GFAP assays for many years, but now that we can dichotomize people by amyloid in early disease stages, we can see its power,” noted Nicholas Ashton, UGothenburg.
Blood Takes It. In BIOFINDER, plasma GFAP levels rise in Aβ-positive groups. CSF GFAP ticks higher in non-AD dementias. [Courtesy of Pereira et al., Brain 2021.]
Delving deeper, Pereira found that plasma GFAP correlated with amyloid PET not only in AD cases, but in all cognitively unimpaired individuals and groups. This fits with Wang’s data suggesting GFAP is an early marker of neuroinflammation in AD. In contrast, CSF GFAP only correlated with amyloid PET in people who were also amyloid-positive as per CSF, and only when they were also cognitively impaired. When calculated voxel by voxel, only plasma GFAP correlated with amyloid, once again suggesting a more sensitive correlation.
As in Wang’s cohort, Pereira, too, found that plasma GFAP tracked Aβ PET SUVRs. When plotting sensitivity over specificity, plasma GFAP predicted PET positivity with an AUC of 0.76, versus 0.69 for CSF GFAP. Both markers predicted cognitive decline in longitudinal analyses, though only plasma GFAP predicted amyloid accumulation.
All these studies used the same Quanterix SIMOA assay for GFAP. This eliminates errors arising from assay or platform variations, said Ashton. “Unlike for p-tau species, there is only the one GFAP assay, so it is really developing consistent result across cohorts,” he said.
Even so, scientists are perplexed by the contrast between plasma and CSF GFAP. The former seems to reflect amyloid accumulation, whereas the latter may reflect neuroinflammation caused by other pathologies. “It is really a surprise to everyone,” said Ashton. But why does only plasma GFAP predict amyloid? “It is counterintuitive,” agreed Michelle Mielke, Mayo Clinic, Rochester, Minnesota. “I have not heard a good reason for it.” Ashton told Alzforum that scientists are focusing on two potential explanations. One is that GFAP degrades faster in the CSF, and may be more sensitive to freeze/thaw cycles, which could skew data from stored samples. Ashton favors this idea, and would like to test if there is a difference between fresh plasma and fresh CSF. Or perhaps GFAP does not enter plasma via CSF, but through some other mechanism, possibly directly from astrocytes into the blood vessels. This idea is plausible because astrocytes are in direct contact with pericytes on small blood vessels of the neurovascular unit.
Indeed, both Ashton and Mielke noted that plasma GFAP might shoot up in cerebrovascular disorders and traumatic brain injury. “We need a comprehensive study that looks at both cerebrovascular and AD pathology, aging, atrophy, and various cognitive domains to fully understand exactly what this assay is measuring and how it may be more useful,” said Mielke. “That said, there are studies that suggest it is more specific to AD neurodegeneration, so that is really promising.”
As for p-tau, that other early marker, Ashton presented the latest at AAIC. Earlier this year, he had seen that plasma p-tau231 was one of the earliest of them all, appearing before p-tau181 and before PET scans could detect amyloid. That data came from four different cohorts, including a small autopsy group of 47 brain donors (Feb 2021 news). Working with Douglas Galasko, University of California, San Diego, Ashton has since added a second autopsy cohort of 312 patients at UCSD’s Alzheimer’s Disease Research Center. These volunteers had had plasma samples taken every other year for decades, including last samples taken within five years of death, and reflect a more heterogenous population.
Similar to the prior data, plasma p-tau231 distinguished AD from other neurodegenerative diseases and strongly correlated with CERAD scores of neuritic plaques and with Braak stage at autopsy. In people who had died at Braak stage V/VI, plasma p-tau231 had risen in plasma at least 10 years prior. Further, plasma p-tau231 best predicted cognitive decline over four years. Similarly, in samples from PREVENT-AD, a Canadian study that tracks cognitively normal people at risk for AD, Ashton found that in those who tested positive for amyloid based on plasma Aβ42/40 levels, plasma p-tau231 predicted subtle cognitive decline over six years.
How about in people with early signs of AD pathogenesis? For this, Ashton turned to the ALFA+ cohort in Spain. Among 384 people, average age 61, who are cognitively healthy and had PET scans for amyloid, plasma p-tau231 predicted Aβ-positivity better than did plasma neurofilament light or plasma p-tau181, but no better than plasma GFAP. However, all tau markers outperformed plasma GFAP in correlating with change in CSF Aβ42/40.
Could this be where plasma p-tau231 shines? “We think this is a robust clinical biomarker that outperforms plasma GFAP and p-tau181 in predicting soluble Aβ changes that occur prior to plaque formation,” said Ashton.—Tom Fagan
Does COVID-19 increase the risk of dementia? While that will take decades to answer, researchers are beginning to unravel how this disease might injure the brain. At the Alzheimer’s Association International Conference held in July in Denver and online, researchers described lingering cognitive deficits and biomarker changes in people six months after COVID infection. Thomas Wisniewski and colleagues at the New York University School of Medicine found lower plasma Aβ42 and higher tau levels in "neuro-COVID" cases than those without brain symptoms. Nelly Kanberg, University of Gothenburg, Sweden, saw plasma neurofilament and glial fibrillary acidic protein rise in the weeks following severe COVID, regardless of neurological problems, then normalize by six months. Likewise, Patrick Smeele and Lisa Vermunt of Amsterdam UMC saw plasma NfL rise then fall during the first few weeks of ICU stays. What this means for long-term dementia risk, if anything, remains unclear.
Biomarker Trajectories
Some people experience cognitive problems and other neurological symptoms during COVID. Could annoying brain fog foretell something more sinister? During the illness, total tau, NfL, and GFAP rise in the cerebrospinal fluid, while the latter two are known to climb in the blood (Jan 2021 news; Espíndola et al., 2021). While most research has focused on hospitalized people, one small study showed an uptick in CSF NfL and GFAP in the mildly sick (Virhammar et al., 2020). This hints that the brain may be under attack. How long might these insults last?
So far, some researchers have followed patients for six months after either testing positive or going home from the hospital. Wisniewski and colleagues measured blood markers in 310 adults with severe COVID after they went home from one of four NYC hospitals. Of those, 158 had had neurological symptoms, aka neuro-COVID, during their hospital stay; 152 did not. Average ages were 70 and 73, respectively; in both groups more than half were men.
After six months, people with neuro-COVID had 1.5-times more total tau and p-tau181, half as much Aβ42, and six times higher total tau/Aβ42 ratios in their plasma. Aβ40 levels were similar regardless of neurological symptoms. Does this mean people with neuro-COVID are at risk for dementia? Only a few cohorts have reported Aβ and tau markers so far, and the jury is still out (see below for commentary). Wisniewski was unavailable for comment
Wisniewski's group also measured markers of neuronal injury after six months. Neuro-COVID cases still had 2.5 times more NfL and 1.5 times more GFAP than people who had had COVID without neurological symptoms. This may indicate lingering damage, especially in the former group.
Did biomarkers return to normal in COVID cases compared to people who never caught the virus? Kanberg and colleagues led by Magnus Gisslén at UGothenburg suggest so in their poster, and a paper published July 29 in EBioMedicine. They recorded neurological symptoms and collected blood from 51 controls and 24, 28, and 48 adults who had had mild, moderate, and severe COVID, respectively, at the Sahlgrenska University Hospital in Gothenburg. Sixty-eight percent were men, average age 56. Kanberg followed them for up to 7.5 months after symptom onset. The scientists analyzed plasma NfL, GFAP, and growth factor GDF-15, also called MIC-1, a marker of organ and vascular brain injury (McGrath et al., 2020).
Compared to controls, mild COVID patients had no change in their plasma markers. Moderately and severely ill people had higher GDF-15 concentrations. As for neuronal injury markers, GFAP levels jumped during the first few weeks of moderate and severe illness, then dropped back to normal over the next few months. While NfL remained steady in moderate cases, it soared during the first two months of severe illness, then dropped to normal by six months (see image below). “This pattern could indicate an initial astrocytic reaction followed by lingering neuronal injury,” Kanberg told Alzforum. The scientists did not measure levels of Aβ40, Aβ42, total tau, and p-tau181 in these samples, though Henrik Zetterberg, also at UGothenburg, said they will.
Settling Down
Plasma NfL (top) and GFAP (bottom) mainly remained steady in mild (left) and moderate (middle) COVID cases but spiked in the severely ill (right). Markers normalized by six months. [Courtesy of Kanberg et al., EBioMedicine, 2021.]
Half of the volunteers in the Gothenburg cohort had neurological problems at six months, though biomarkers did not correlate with thise. “NfL and GFAP normalized in every patient, regardless of whether they had long-term symptoms or not,” Kaj Blennow, U Gothenburg, told Alzforum. Kanberg and colleagues concluded that long-term neurological problems from COVID do not indicate ongoing CNS injury or neurodegeneration.
Smeele, Vermunt, and colleagues led by Charlotte Teunissen at Amsterdam UMC measured an NfL upsurge in blood taken from 31 adults who had severe COVID and were admitted to the university’s medical center. The researchers took blood samples from the volunteers every few days for up to four weeks. The average age was 63, 70 percent were men. Within 90 days of admission, seven had died. In survivors, plasma NfL levels peaked around 2½ weeks after admission into the ICU; in those who died, plasma NfL levels kept climbing for at least 3½ weeks (see image below).
Diverging Paths
Logarithmic spaghetti plots of individual COVID patients show plasma NfL peaked sooner, and at lower levels, in survivors (thick yellow line) than those who perished (purple line). [Courtesy of Patrick Smeele, Amsterdam UMC.]
Likewise, Meredith Hay, University of Arizona, Tucson, and colleagues reported higher serum NfL in COVID ICU patients than in people who were there for other reasons, see preprint posted May 2 on medRxiv (Hay et al., 2021). Of 100 people admitted to the university hospital, 89 had COVID. On average, people with the virus had 12 times the amount of NfL in their blood as those admitted for other reasons, 230 pg/mL in COVID ICU vs. 19 pg/mL. The level was even higher in people with COVID who also had cardiovascular disease and diabetes. The authors thought this could mean that widespread inflammation in the body could fuel the neuronal damage seen in COVID, and hint at whose cognition stays at risk later in life.
COVID and Cognition
People have reported neurological symptoms after contracting the virus, with some persisting for weeks or months (Apr 2021 conference news). How common are these?
Wisniewski and colleagues found neurological disorders in 13.5 percent of 4,491 adults hospitalized with COVID in New York City (Frontera et al., 2021). The researchers phoned up 382 survivors six months later to follow up. Half were still struggling performing daily activities, or their cognition as judged by the Montreal Cognitive Assessment had declined. People who had had neuro-COVID struggled more with activities and disability, per the modified Rankin Scale, but their cognition had weakened similarly to COVID cases who had not had neurological symptoms (Frontera et al., 2021). Wisniewski drew on this cohort for the biomarker analysis.
For their part, scientists led by Yan-Jiang Wang and Juan Liu at the Third Military Medical University, Chongqing, China, detected cognitive deficits in 10 percent of 1,301 non-severely and 238 severely ill COVID patients older than 60 from Wuhan (Liu et al., 2021). Six months after being discharged from the hospital, COVID cases scored worse on the Telephone Interview of Cognitive Status-40 (TICS-40) than did the 466 uninfected controls. Non-severe cases and controls scored similarly: 5 percent were in the MCI range and less than 1 percent in the dementia range. Severely ill people scored worse, with 25 percent in the MCI range and 11 percent in the dementia range.
At AAIC, Gabriel de Erausquin, University of Texas Health, San Antonio, presented striking results from an Argentinian cohort of 330 COVID cases, ranging from mild to severe. Four to seven months after testing positive, researchers assessed participants using the global Clinical Dementia Rating scale and tests to gauge memory, executive function, orientation, and language. While no one in this cohort had complained of having memory or cognitive issues before COVID, 60 percent did after the infection. “This is 10 times the prevalence of impairment in the older general population,” de Erausquin said. While one-third of those participants scored poorly in one cognitive domain, the remaining two-thirds struggled in multiple domains.
For Göran Hagman, Miia Kivipelto, and colleagues at the Karolinska University Hospital in Sweden, COVID outcomes were less clear. They tested 32 adult COVID survivors three months after they were discharged from the ICU. Despite several participants expressing concern about their cognition, testing showed no pattern. “Some patients were cognitively intact, but others had deficits in attention and executive function,” Kivipelto told Alzforum.
The researchers did find signs of microvascular injury via structural MRI. They are now asking if this injury relates to cognitive problems. “We want to learn if brain damage and poorer cognitive scores match with the subjective cognitive decline people are reporting,” Kivipelto said. To figure out what is going on at a deeper level, Zetterberg and de Erausquin plan to measure neuronal injury and neurodegeneration markers in blood samples taken from the Swedish and Argentinian volunteers.
Into the Unknown
Many people fear long-term effects from contracting COVID, especially brain fog and other neurological consequences. What do the spikes in the markers measured so far mean for future dementia risk? Zetterberg found it reassuring that the markers normalize within six months. “I feared that SARS-CoV-2 would induce ongoing neuronal injury,” he told Alzforum.
Then what causes the neurological symptoms of long COVID? “Lasting symptoms may indicate lingering brain network disturbance induced by the virus rather than frank CNS injury,” said Zetterberg, who added that other infections can leave a person with brain fog for months without evidence of simultaneous brain injury.
Other researchers thought that how severely ill a person is, not COVID itself, may predict dementia risk. “Hospitalization, in general, is a risk factor for dementia, but we do not know why,” Vermunt said. “How sick you get and how you recover may predict who is at risk of future dementia,” Smeele added. Kanberg agreed, noting that the rise in NfL and GFAP in severe COVID cases may be prognostic. “This may mean they are prone to future neuronal injury, so we could track these people to see if they have any long-term problems or develop dementia,” she said.
Vermunt wondered if the opposite might be true—that people who already have prodromal dementia are more likely to have worse COVID. “Perhaps this was why they developed severe disease, but that needs to be answered with population-based research,” she said.—Chelsea Weidman Burke
Frontera JA, Sabadia S, Lalchan R, Fang T, Flusty B, Millar-Vernetti P, Snyder T, Berger S, Yang D, Granger A, Morgan N, Patel P, Gutman J, Melmed K, Agarwal S, Bokhari M, Andino A, Valdes E, Omari M, Kvernland A, Lillemoe K, Chou SH, McNett M, Helbok R, Mainali S, Fink EL, Robertson C, Schober M, Suarez JI, Ziai W, Menon D, Friedman D, Friedman D, Holmes M, Huang J, Thawani S, Howard J, Abou-Fayssal N, Krieger P, Lewis A, Lord AS, Zhou T, Kahn DE, Czeisler BM, Torres J, Yaghi S, Ishida K, Scher E, de Havenon A, Placantonakis D, Liu M, Wisniewski T, Troxel AB, Balcer L, Galetta S.
A Prospective Study of Neurologic Disorders in Hospitalized Patients With COVID-19 in New York City.
Neurology. 2021 Jan 26;96(4):e575-e586. Epub 2020 Oct 5
PubMed.
Frontera JA, Yang D, Lewis A, Patel P, Medicherla C, Arena V, Fang T, Andino A, Snyder T, Madhavan M, Gratch D, Fuchs B, Dessy A, Canizares M, Jauregui R, Thomas B, Bauman K, Olivera A, Bhagat D, Sonson M, Park G, Stainman R, Sunwoo B, Talmasov D, Tamimi M, Zhu Y, Rosenthal J, Dygert L, Ristic M, Ishii H, Valdes E, Omari M, Gurin L, Huang J, Czeisler BM, Kahn DE, Zhou T, Lin J, Lord AS, Melmed K, Meropol S, Troxel AB, Petkova E, Wisniewski T, Balcer L, Morrison C, Yaghi S, Galetta S.
A prospective study of long-term outcomes among hospitalized COVID-19 patients with and without neurological complications.
J Neurol Sci. 2021 Jul 15;426:117486. Epub 2021 May 12
PubMed.
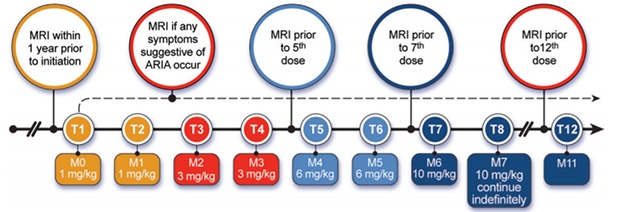
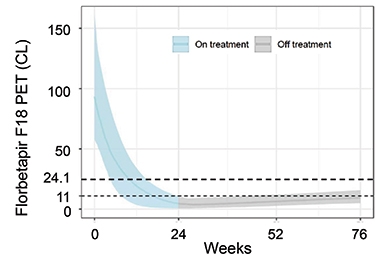
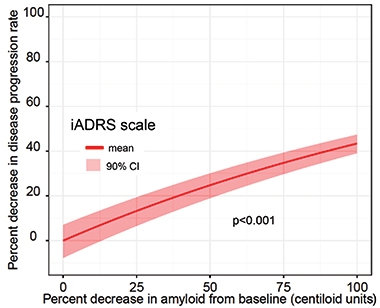

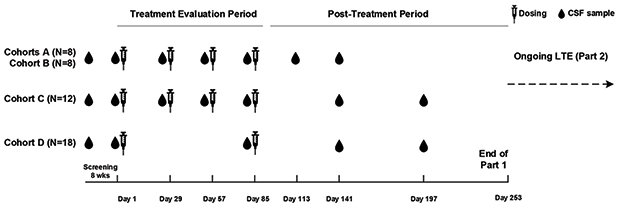




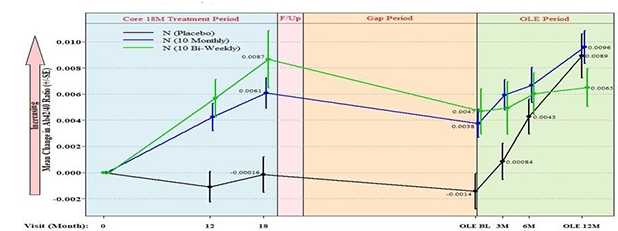









Comments
No Available Comments
Make a Comment
To make a comment you must login or register.