Held in Boston’s Park Plaza Hotel, this year’s CTAD conference filled its chosen venue to fire regulation capacity. The space bustled with scientists heading to talks, posters, and various private meetings in rooms up and down the hallways. Despite a notable absence of success stories for large trials, a sense of optimism pervaded the scene, which stands in marked contrast to a public perception of all-failure-all-the-time in Alzheimer disease trials. True, verubecestat data was flat-out negative in mild to moderate AD and some scientists began to fret about a possible class effect, but on the other hand, several anti-Aβ antibodies posted evidence of removing amyloid plaque reasonably safely from the human brain, and all breaks were off on prep work for multiple large-scale prevention trials in the preclinical phase of this long disease. CTAD news ran the gamut from creative approaches to developing drugs (think young men’s plasma fraction) to creative approaches to measuring subtle cognitive change (think burst mobile tests and digital pen), to on-the-spot ApoE genotyping (think cube). Learn the latest with our 13-part news series.
10th CTAD: Finally, Alzheimer’s Field Is Serious About Prevention Trials
To attend the 10th Clinical Trials on Alzheimer’s Disease conference was to witness a field at large grapple with the realization that its focus is in the midst of swinging downward from the visible tip of the iceberg that is Alzheimer’s toward its underwater bulk. Gathered in Boston November 1 to 4, trialists were training their eyes squarely on the 15 years of pathogenic buildup that lead to the roughly eight years of symptomatic disease, which scientists increasingly refer to as brain “organ failure” or “end-stage.”
To be sure, CTAD featured trial data on both amyloid- and non-amyloid drugs, as it does every year, and some of those trials were in mild-to-moderate disease (e.g. see Part 6, Part 8, Part 12 of this series). But the field’s main thrust clearly was on how to apply a new understanding of the pre-dementia stages of AD toward running better therapeutic trials.
“The great advance over the past 10 years since CTAD began is that we now know that AD really is a continuum,” said Reisa Sperling of Harvard Medical School in Boston. “It begins well before what we recognize clinically as dementia and even before the stage we recognize as prodromal. So the question has become: How can we treat the presymptomatic stage? This may be our best opportunity to bend the curve toward normal aging.”
Change would come not a day too soon. The abysmal success rate of the past decade’s clinical trials has been publicized in overly simple terms, said Rachelle Doody, Roche/Genentech, Basel, Switzerland. A public perception of failure clings to Alzheimer’s trials. It must change to one of learning and hope in order to help rally the thousands of volunteers, and motivate site staff, for the massive prevention initiatives that are gearing up around the world (see Part 13 of this series).
After years of debate about causes and clinical groupings, the field’s pulse now is felt most strongly in efforts to identify, engage, and characterize asymptomatic, at-risk participants (see Part 5 of this series). Innovation focuses on ways to tighten up variability, and to capture richer data sets when measuring a person’s subtle cognitive slide toward dementia, both as disease takes hold and in response to investigational drugs (see Part 4 of this series). Clinically certified CSF tests, as well as up-and-coming research-grade blood tests, are energizing international efforts to bring an early diagnosis into routine clinical care and to become more efficient at screening large populations for suitable trial participants (see Part 3 of this series). The field’s collective shift toward treating earlier stages of AD requires change at every step of the clinical trial endeavor, from cohort-building to outcome measures and statistical methods. Advances on display at this year’s CTAD splashed across all these fronts.
Measuring Early Decline
For starters, consider this question: Is it possible to track early cognitive decline? Yes, said Sperling, citing data from the Harvard Aging Brain Study as an example. As shown in previous observational cohorts of cognitively normal aging people, HABS participants with brain amyloid deposition do decline in subtle ways, and this can be measured with the Preclinical Alzheimer Cognitive Composite, aka PACC, a new battery of composite tests. In contrast, HABS participants without brain amyloid get better on the PACC as they retake the tests on subsequent visits. This divergence between loss and learning shows up on each component of the PACC, and it grows over time, Sperling said.
Next, is it possible to measure preclinical progression with biomarkers? Yes again, said Sperling. The model is that preclinical AD represents a confluence of cortical amyloid deposition and tau pathology expanding beyond its age-related confines in the medial temporal lobe into the cortex. This model is being tested by multimodal PET studies that track both amyloid and tau over time, and it appears to hold up. “Preclinical AD is amyloid plus tau spreading into cortex at Braak stages III to IV,” Sperling told the CTAD audience.
Do cognition and pathology relate to each other at this stage? Once again, the answer is yes. Even within the narrow cognitive range of HABS participants, those with the most brain amyloid and the most tau score lowest on the PACC. “In people with amyloid, tau drives memory decline. So if you have a lot of amyloid and a lot of tau, you will not stay normal for long,” Sperling said.
Narrow as it is initially, this gap soon widens. At CTAD, Bernard Hanseeuw of Massachusetts General Hospital, Boston, told of a man who was clinically normal at baseline despite having cortical amyloid plus tau in his entorhinal cortex. Eighteen months later, tau had spread but his MMSE, free recall, and PACC held steady; alas, another year later, tau had invaded the inferior temporal cortex on both sides and his PACC turned abnormal. This pattern holds true as HABS collects longitudinal measurements, Hanseeuw said. To date, serial results on cognition, amyloid, and tau are available for 60 HABS participants. The key finding is that an increase in tau PET in the temporal cortex better predicts decline in cognition than do amyloid or even baseline tau (for details, see Part 2 of this series). “Tau PET is a promising marker for monitoring disease progression and drug efficacy,” Hanseeuw said.
This is more than one enthusiast’s opinion. After a slow start, and problems with some initial investigational tracers, drug developers across the globe are now closely watching validation studies of newer tracers and starting to use them in their trials. “We all in the field see this. Tau PET will be an outcome measure, together with more sensitive cognitive composites. Amyloid is better as an inclusion criterion,” Marc Cantillon, who is advancing an anti-Aβ oligomer antibody into Phase 2 for the Japanese company Kyowa Kirin, told Alzforum (see Part 10 of this series).
Talks throughout CTAD strengthened the perception that tau PET matches cognition and may pack a punch in drug trials. Sergey Shcherbinin, Eli Lilly and Company in Indianapolis, showed an intriguing parallel between tau and cognition when he presented his refinement of an existing approach to derive continuous, stereotypical biomarker staging diagrams from short-term serial data collected in observational study cohorts (e.g., Villain et al., 2012; Villemagne et al., 2013; Jack et al., 2013). Statisticians are honing methods to transform data on the relationship between a given biomarker’s baseline level and its annual rate of change—typically garnered in three- to seven-year stretches of observation in various cohort studies—into sweeping curves that depict the entire 20-year pathogenic process. Ideally, the curves in such an improved, data-driven staging model should incorporate the relationship between individual biomarkers, Shcherbinin said at CTAD.
To that end, Shcherbinin tried his method on all ADNI data available as of February 2017. His way of deriving a trajectory from baseline and rate-of-change data yielded three different curve shapes for the five markers measured. Florbetapir PET assumed a flat sigmoid shape; FDG PET and hippocampal volume both assumed roughly linear shapes, reflecting constant rates of change. However, in line with Hanseeuw’s data, the curves for two markers assumed a parallel, exponential shape marked by accelerated change with higher baseline levels of abnormality. They were cognition and tau PET.
In toto, a picture is emerging of an exponential increase of tau, followed by cognitive decline, in the presence of amyloid. To many scientists, this strengthens the view that once amyloid reaches its plateau, the disease process becomes independent of amyloid.
Targeting Early Decline in Therapeutic Trials
“How can we use this information to help us test the right target with the right drug at the right stage?” Sperling asked. CTAD featured updates on some of the secondary prevention trials that are already underway and additional trials on the drawing board. As currently designed, AD prevention trials can last up to a decade from planning to data. To incorporate research on drugs and biomarker staging that is newly coming out during those years, scientists adjust ongoing trials as regulators allow. For example, after solanezumab flopped in Phase 3 for mild AD, the A4 solanezumab prevention trial in people who are amyloid-positive but cognitively normal quadrupled the dose to 1.6 grams of solanezumab infusion every month. A4 is also lengthening the treatment period to five years in hopes that these changes will reveal what is expected to be a small treatment benefit.
Ditto for the API prevention trial of crenezumab in presenilin-1 E280A mutation carriers in Medellin, Colombia, and for the DIAN-TU prevention trial of gantenerumab in autosomal-dominant AD mutation carriers. Both studies have increased the dose midway through, as have the prodromal and mild AD trials on these antibodies in late-onset AD (see Part 7 in this series).
Incidentally, these dose increases jibe with the newest results on the anti-Aβ antibody aducanumab. At CTAD, Biogen’s Samantha Budd showed 36-month treatment data from the ongoing long-term extension of the aducanumab Phase 1b PRIME trial of people with mild AD. In people who are receiving the highest dose, 10 mg/kg, brain amyloid deposition had dropped below SUVR 1.1, the preset threshold of positivity, by 80 weeks, and stayed this low at 166 weeks. This does not mean plaques are completely gone, Budd said in response to a question, but rather, that their overall burden has dropped below a brain-wide cutoff.
To mitigate aducanumab’s ARIA side effect, Biogen is evaluating a titration scheme that gradually approaches an effective dose. At CTAD, Philipp von Rosenstiel of Biogen showed long-term-extension data for this group of 18 participants. After 24 months, the titration group had about the same amount of amyloid reduction as the 6 mg/kg fixed dose group, and comparable safety. Biogen uses titration in its ongoing Phase 3 program of aducanumab in mild AD.
In toto, both prevention and symptomatic trials of the major antibodies—aducanumab, gantenerumab, crenezumab, solanezumab—are pushing in the same direction of giving high doses for long periods of time.
Further adapting to emerging science, prevention trials have started to add tau PET. A4 added flortaucipir in 2016 and has scanned 381 participants so far. Baseline results mirror Hanseeuw’s finding in the HABS natural history cohort, in that half of these cognitively normal people with amyloid already have cortical tau in their inferior temporal lobes, posterior cingulate, and precuneus. Once again, tau was bad news even in this ostensibly normal phase. A4 tightly limited cognitive inclusion criteria, such that participants had to be normal on MMSE, within one standard deviation on other tests, and could not be supernormal. “Even within this narrow cognitive band, people with highest amyloid have the most tau, and people with the most tau have the lowest memory scores. This was striking to me,” Sperling said. Given this distribution, some scientists speculated that perhaps the less-advanced people within this A4 cohort may still derive a benefit from an antibody that reduces monomeric Aβ, whereas people whose tau is spreading may not, because their disease has become independent of amyloid.
Sperling is not waiting for the answer. “When we submitted the A4 grant five years ago, prevention trials were considered fantasy. Now we know they are feasible, and we are already learning screening data in cohorts who are at risk genetically or by biomarkers. But we cannot wait for results from current trials; we must start additional trials with multiple mechanisms now,” Sperling said. To that end, the A5 trial, aka EARLY, is evaluating Janssen’s BACE-1 inhibitor JNJ-54861911 in a slightly younger population starting at age 60 (instead of A4’s 65), who have amyloid accumulation either by CSF or PET. Fourteen sites are currently enrolling. The Novartis/Banner Generation trials of the BACE inhibitor CNP520 and the active Aβ immunotherapy CAD106 are also enrolling cognitively normal people starting at age 60 who are at genetic or biomarker risk of developing Alzheimer’s dementia.
Alas, ultimately, even age 60 with established amyloid deposition represents a later stage in the long pathogenic process than when Sperling wants to deploy anti-amyloid drugs. Therefore, the next trial, A3, or Ante-Amyloid Prevention of AD, is going to search for people aged 50 to 75 who are at high risk of accumulating amyloid and developing the subsequent biomarker changes predicted on AD staging diagrams. Unlike A4 and EARLY, which measure success on the PACC, this four-year trial will use biomarkers as primary outcomes and cognition only as an exploratory outcome. Alternatively, A3 could pair up with a sister trial of the same drug in a later-stage preclinical population in whom cognitive change is more robustly measurable within the timeframe of a trial, Sperling said. A3 is NIA-funded as part of a public-private partnership, and finalizing its choice of drug now. Who could enter A3? For one, people who tried to join A4 or EARLY but fell below the amyloid cutoff. At CTAD, Sperling noted that 529 people who failed amyloid scanning in A4 have already joined the observational LEARN study as a springboard for an earlier-stage treatment trial that will accept them. For another, A3 will draw people from the TRC PAD cohort being built by the Global Alzheimer’s Platform, or the EPAD-TRC, a trial-ready cohort being built by the European Prevention of Alzheimer’s Dementia project (Aug 2016 conference news), or other online or local registries.
In the big scheme of things, the A3 study represents a link in the needed chain of trial evidence researchers are building to support their push for drug evaluation into ever-earlier disease stages, until they are able to pull off primary prevention of amyloid buildup in middle-aged people. The DIAN-TU is already working on this for people who carry autosomal-dominant mutations (Aug 2017 conference news); A3 is a necessary step toward primary prevention of LOAD. “We are moving toward the left on the staging diagrams,” said Bruno Dubois from University Salpêtrière Hospital in Paris.
How to Fill Prevention Trials?
The advent of prevention trials and their prodigious recruitment needs are spurring attempts at speeding things up. Consider the A4 trial. It started enrolling in early 2014. By November 2017, 67 A4 sites were active in North America, Australia, and Japan. The good news is that A4 will slightly exceed its target by the time recruitment closes at the end of 2017; however, to find these 1,160 participants, sites had to screen nearly 7,000 people, 4,480 of them with an amyloid scan. Of those, only 30 percent were eligible for A4, Sperling reported at CTAD.
Several years to fill a single early stage trial simply does not seem acceptable any more, and experiments to accelerate recruitment are happening throughout the field. At CTAD, Chris Rowe of Austin Health in Melbourne, Australia, presented an example targeted at prodromal-stage trials. “Our new project addresses the problem that many people are being diagnosed clinically every day, but do not get referred to trials,” he told the audience. Rowe’s site has been doing amyloid PET scans since 2004, and amyloid PET is well-known in Australia but not covered by insurance. What if he could give free amyloid scans to referring clinicians across Melbourne for patients who appear eligible for trials and interested in joining one? In this scheme, a trial sponsor pays for the scans done at Rowe’s center. It is a win for all sides, Rowe said, because the sponsor gets prescreened candidates and reduces the trial’s screen failure rate; the clinician can offer a better diagnosis and some hope when telling his or her patients they have early AD; and the patient gets tangible options for fighting the disease by being pointed toward open clinical trials.
AstraZeneca funded the project, and Rowe’s center fielded referral candidates for scanning who fit the inclusion criteria of AstraZeneca/Lilly’s AMARANTH Phase 3 trial of the BACE inhibitor lanabecestat in early AD. However, referring clinicians were not bound to recommend only this trial to their patients once the scan result was in.
Patients referred from across Melbourne, a city of 4 million, were prescreened by phone and then in person at the city’s Florey Institute of Neuroscience and Mental Health. Over the past 18 months, 64 clinicians referred 342 patients. Of those, 108 failed the telephone screen and 82 failed the on-site screen. Of the 150 who got a PET scan, 115 were positive for amyloid plaques, Rowe said. Of those, the Amaranth trial accepted 41.
Why did half still not get in after that much prescreening? Fourteen people declined when they saw how complex the trial was. Ten had deteriorated, 13 had medical reasons or were on disallowed medications. “I am concerned by these exclusions,” Rowe told the audience. Three more people who had positive NAV4694 scans were negative on a subsequent florbetapir scan done as part of Amaranth. “This is not surprising because florbetapir is less sensitive than NAV4694,” Rowe said (May 2010 news).
An ongoing extension of the program referred 27 additional people to Amaranth. Of the original 115 with positive scans, 32 checked out trials other than Amaranth and 26 of those enrolled. Overall, the program dramatically exceeded the recruitment targets of all the Melbourne AD trial sites, Rowe said. The sponsor’s cost per enrolled person was US$12,000. Importantly, every newly diagnosed patient took up the offer to enter a drug trial. “When people see their scan and hear a diagnosis, they really want to do something about it,” Rowe said.
“This is practical and extremely helpful,” said Steve Salloway of Butler Hospital in Providence, Rhode Island. It could be replicated at other PET centers that have a strong referral base among AD clinicians.
To bring down recruiting time, clinicians want quick tests to tip them off about the likely presence of brain amyloid in a given person; to rein in cost, they specifically want prescreening tools that help them decide who to refer for a PET scan. Besides fluid-based Aβ tests, there may soon be other ways of going about it. John Hardy of University College, London, started his CTAD keynote by saying, “As geneticists, we have not yet done anything terribly useful for patients.” This may soon change, however, which is why Hardy addressed an audience of trialists. He believes that polygenic risk scores, aka PRS, have become good enough to reduce the screen failure rate of prodromal and prevention trials.
In the current era of GWAS and large-scale sequencing, neurogenetics has reached a point where it can apply to clinical use the collective set of variants gathered for the nearly 40 genes that are universally accepted to influence AD risk. The time is right because most genetic variance has been captured. “We know that not all monozygotic twins are concordant for AD, so we will never be able to use genetics alone to predict absolutely who will get AD,” Hardy said. But polygenic risk scores derived from GWAS variants come close, predicting up to 90 percent of genetic risk, much as twin studies do (Escott-Price, Shoai, et al., 2017; Escott-Price, Myers et al., 2017). Indeed, at CTAD, UCL’s Maryam Shoai presented a study testing a new polygenic risk score derived from a training set of 1,700 neuropathologically confirmed cases. This PRS predicted with 90 percent sensitivity who was amyloid-positive among a second set of 600 ADNI samples, Shoai showed. Also at CTAD, Chin Hong Tan in Rahul Desikan’s group at University of California, San Francisco, presented similar data on a genetic tool they call polygenic hazard scores.
Such a test could serve as an upstream check of who should go on to have a PET scan as part of trial screening. It could also become part of the characterization of at-risk participants in the trial-ready cohorts being built in Europe, North America, and Australia. “You can use this now to cut down on PET scans and lumbar punctures,” Hardy told the assembled trialists in Boston.
Perhaps more provocatively, polygenic risk scoring can also unmask misclassification in general clinical samples, Hardy told the audience. For example, applying PRS to the samples that formed the basis of the large IGAP GWAS showed that 10 percent of its E4 carriers and 30 percent of noncarriers were classified as healthy controls when their PRS indicated that they were likely amyloid-positive. At age 75, one in five were clinically classified as controls even though their genetics indicated they had brain amyloid deposition, Hardy said.
To Paul Aisen, University of Southern California, the improving AD biomarker staging scheme, combined with tools to capture risk and biomarker status of people in standing trial cohorts, suggests a game plan for the future. “For primary prevention, we will use a BACE inhibitor. I think it will be a cure. For secondary prevention, we will use an antibody and a BACE inhibitor. For prodromal AD, we need a third drug of a different class, probably a tau drug. For mild to moderate AD, I don’t know yet what the combination will be. We need to learn more. But we will succeed,” Aisen told Alzforum.
To this end, scientists at CTAD renewed their call for industry and regulatory scientists to buckle down and start combination trials of two investigational drugs. Word in the hallways had it that some of the companies that have both the necessary Aβ antibodies and BACE inhibitors drugs—Lilly, Roche, and Eisai, for example—are nearing the launch of a first such trial in early 2018, though no one was ready to make an announcement yet.
Meanwhile, a range of single-drug trials are moving forward (see Part 9, Part 11 of this series), while others closed the book on their candidate drugs (see Part 12 of this series).—Gabrielle Strobel
At CTAD, Tau PET Emerges as Favored Outcome Biomarker for Trials
At the Clinical Trials on Alzheimer’s Disease meeting held November 1–4 in Boston, tau PET emerged as the crowd favorite among candidate biomarkers for measuring progression, in both natural history studies and future trials. It’s no news that tau PET tracks neurodegeneration and cognitive decline in AD better than amyloid PET. Now, researchers are pinning down details of how the tau signal changes over time and how that change relates to symptoms. These are two critical bits of information for gauging the method’s usefulness as a biomarker for prevention and treatment trials. A new PET ligand, PI-2620, offers an improved alternative to the first-generation tracer flortaucipir. With low background, PI-2620 binds multiple tau isoforms, bringing PET to non-AD tauopathies. Separately, an attempt to visualize a different aspect of Alzheimer’s—a ligand that directly measures synapses—showed losses in the hippocampus that appeared to correlate with cognitive trouble.
To relate tau PET to disease progression, Bernard Hanseeuw of Harvard Medical School and Massachusetts General Hospital, Boston, has been studying participants in the Harvard Aging Brain Study (HABS) over time. At CTAD, he showed results on 60 people, median age 75. All were cognitively normal when they received baseline PiB and flortaucipir scans for amyloid and tau. They had two more scans after one and two years, and cognitive testing out to three years thus far. Hanseeuw analyzed changes in tau in their temporal neocortices.
In this group, Hanseeuw found that tau increased faster than amyloid. It would take 20 years for amyloid deposition to go up by one standard deviation, but only five years for tau. Tau PET was also the more consistent measure, showing less variability from person to person than amyloid. Both amyloid and tau deposition accelerated with time, such that the annual increase was greater in people who started out higher at baseline.
How did that relate to cognitive decline? How fast tau accumulates best predicted cognitive decline. This measure outperformed PiB accumulation, baseline PiB, or baseline tau. Even in people with high baseline amyloid and tau, cognitive decline correlated better with tau than amyloid. And while baseline PiB or tau themselves indicated future cognitive decline, the rate of tau accumulation was the stronger predictor. The results need to be validated in a larger sample with longer follow-up, Hanseeuw said. Even so, he concludes that tau PET “looks like a promising biomarker for monitoring disease progression and drug efficacy in AD trials.”
Echoing the HABS findings, Reisa Sperling, Brigham and Women’s Hospital, Boston, reported that tau is a sensitive marker for preclinical cognitive decline in the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s trial cohort. A4 participants are cognitively normal with elevated amyloid. Baseline flortaucipir PET revealed that about half started the study with increased tau. The higher a person’s amyloid, the more tau they have in the medial and inferior temporal lobes and also in the posterior cingulate and precuneus areas. Sperling said for a cognitively normal group, this was striking. “In A4, we restricted the inclusion criteria to a narrow cognitive range Participants had to be normal on the MMSE, within one standard deviation of normal on other tests, and could not be supernormal. Even within this narrow band, the people with highest amyloid have more tau, and those with the highest tau have the lowest memory scores,” Sperling said.
“These data really highlight that tau PET imaging is a main outcome measure we should consider in clinical trials,” said Oskar Hansson of Sweden’s Lund University. Hansson had previously followed observational cohorts with fluid biomarkers for many years, but at CTAD he offered more validation of flortaucipir in AD from the Swedish BioFinder and other studies. Hansson compared flortaucipir PET with cerebrospinal fluid tau. CSF tau increases in late preclinical stages, when people are still cognitively normal, but then shows little further change as symptoms progress. Hansson said tau PET correlates only moderately with CSF tau, but tracks both brain atrophy and cognitive symptoms well.
Hansson calls CSF tau a “state” marker, which signals that someone is developing AD, while tau PET serves as a staging marker, which gives information about how far along they are.
On the down side, flortaucipir has performed poorly in non-AD tauopathies. The ligand binds only weakly to four-repeat (4R) tau isoforms, like those that accumulate in corticobasal syndrome (CBS) or progressive supranuclear palsy (PSP). In Hansson’s recent work, flortaucipir was able to identify pathology in the correct brain regions in CBS, but the signal was much lower than in AD (Smith et al., 2017). In PSP, tau neuropathology correlated with FDG but not tau PET (Smith et al., 2017). Overall, Hansson concluded that flortaucipir might be good enough to identify pathology in CBD, but not PSP.
Tracking Tau. PI-2620 in five people who are positive for amyloid deposition measured by florbetaben. Tau PET signal strength reaches up to 3.5 (see scale on right) in some areas; the person on the far right was borderline amyloid-positive. [Courtesy of Andrew Stephens.]
Beyond Flortaucipir, What Else Is There?
In part because flortaucipir is not widely available, or is too expensive for some groups, at least five new tau tracers have come into development (Apr 2017 conference news). At CTAD, Andrew Stephens, Piramal Imaging, Berlin, showed off his company’s entry, PI-2620. The compound binds both the three-repeat (3R) and 4R tau isoforms, and recognizes neurofibrillary pathology in AD, Pick’s disease, and PSP. Stephens showed robust uptake of PI-2620 in a patient with PSP, with binding to the bilateral globus pallidus and substantia nigra. Quantitating the PET signal in cortical and subcortical regions clearly distinguished PSP from AD. “This is a good opportunity for us,” Stephens said.
PI-2620 rapidly enters the brain, peaks at 60 to 90 minutes, and leaves the brain. After dosing, PI-2620 completely clears the body without producing lipophilic metabolites that re-enter the brain and muddy the measurement, an occasional problem in PET tracer development. Its effective dose is comparable to that of other tracers, Stephens said, allowing for two to three scans per year while staying within radiation exposure limits. Stephens emphasized very low off-target binding, with no signal in the choroid plexus, striatum, amygdala, and other midbrain structures where other tracers have run into trouble. “Choroid plexus binding masks hippocampal signal, so getting rid of that is a significant advantage,” Stephens said. He sees no uptake in elderly controls, but sees a typical distribution, distinct from amyloid, in AD patients. Regional standardized uptake volume (SUVr) values in abnormal regions reached 3.5—a stronger signal than some other tau tracers. PI-2620’s test-retest reproducibility was good in healthy controls and AD, he said.
Trial Ready? Piramal’s PI-2620 shows a typical tau distribution pattern with varying degrees of accumulation in three amyloid-positive people with probable AD enrolled in the Proclara NPT088 trial. [Courtesy of Andrew Stephens.]
PI-2620 will be tested in several clinical trials, Stephens said at CTAD. The scans will serve as exploratory endpoints in a Phase 1b trial of NPT088, an investigational drug from Proclara Biosciences, Cambridge, Massachusetts. A fusion between human immunoglobulin and a bacteriophage capsid protein, the drug is a second-generation version of a prior bacteriophage therapy known to dissociate amyloid aggregates (Levenson et al., 2016; Jan 2013 conference news). In three trial participants scanned thus far with MMSEs between 25 and 19, baseline PI-2620 uptake ranged from minimal to dramatic. Stephens declined to say which other therapy trials are using PI-2620.
Tau en le Cervau.
In these black-and-white scans, MK-6240, a tau PET tracer developed by Merck and now Cerveau Technologies, shows no uptake in healthy controls but extensive uptake in cortical areas in a person with Alzheimer’s. [Courtesy of Christopher Rowe.]
PI-2620 is not the only new commercial tau tracer. At previous meetings, Merck researchers debuted that company’s tau PET ligand, MK-6240, which is progressing through Phase 1. Because Merck develops PET tracers for in-house use but is not a radiopharmaceuticals company, in January 2017 Merck licensed the probe to Cerveau Technologies, a partnership between Enigma Biomedical Group of Toronto and Sinotau Pharmaceutical Group in Beijing. Backed by a consortium of pharmaceutical companies, Cerveau is managing the development of MK-6240 and establishing distribution centers worldwide so it can be used in clinical and academic trials. Cerveau moved fast, and by March had secured a manufacturer and initiated research collaborations with Pedro Rosa-Neto at McGill University in Montreal, Sterling Johnson at University of Wisconsin-Madison, and Christopher Rowe at Austin Health in Melbourne, Australia, all of whom are now studying the tracer in people.
No data on MK-6240 were presented at CTAD, but just prior to the conference, Cerveau announced a partnership with the DIAN-TU at Washington University, St. Louis. Academic groups that will use the probe include Banner Health, Phoenix, New York University, the Clinical Imaging Research Center in Singapore, Massachusetts General Hospital, New York City’s Columbia University Medical Center, Houston Methodist Hospital, VU Medical Center in Amsterdam, and the Centre for Addiction and Mental Health (CAMH) in Toronto, as well as the pharmaceutical companies Biogen, AbbVie, Janssen, and Lundbeck. Rowe told Alzforum that more than 100 human studies have now been done. Thus far, the tracer shows high binding in people with AD, but no non-specific or off-target background uptake in the brain. The tracer will be in multiple clinical trials in 2018, according to Rowe.
What About Synapses—When Can We See Them?
A different substrate of Alzheimer’s disease—synapses—has traditionally lacked in vivo human biomarkers, even though researcher have long known that synaptic loss is a major structural correlate of cognitive impairment. In the CSF, this gap is starting to fill with the emergence of neurogranin, and PET may be next.
Synaptic Snapshot.
11C-UCB-J reveals reduced SV2A binding in a patient with AD (right) compared to an older control (left). [Courtesy of Christopher van Dyck.]
Historically, glucose metabolism, measured by FDG-PET, has provided an indirect measure of neuronal activity. Now, a new PET ligand for the synaptic vesicle glycoprotein 2A (SV2A) promises to measure synaptic density more directly. At CTAD, Christopher van Dyck, Yale University, New Haven, Connecticut, presented initial work with 11C-UCB-J in 10 people with AD. Developed by Richard Carson at Yale and UCB Pharma in Belgium, this probe quickly enters the brain, where it binds predominantly gray matter. In people with epilepsy, the compound can detect areas of synaptic loss near the focal point of seizures (Jul 2016 news; Finnema et al., 2017).
In the new work, van Dyck, Ming-Kai Chen, and colleagues scanned 11 PiB-negative, cognitively normal controls and 10 people who were PiB-positive, including five with mild dementia and five with amnestic MCI, in a high-resolution research scanner. They focused on the medial temporal lobe. They expected that early degeneration of entorhinal cortical projecting neurons might thin out the synapses of these cells. Also, hippocampal SV2A protein reduction has been documented in postmortem AD brains (Braak et al., 2011; Robinson et al., 2014).
Indeed, participants with AD had an average 41 percent reduction in tracer binding in the hippocampus. A reference region, the centrum seminovale, did not differ between the groups. The pattern of reduction in 11C-UCB-J differed from FDG-PET, which reflects more widespread decline in synaptic activity in the temporal, parietal, and posterior cingulate regions. In this group of participants, the loss of hippocampal binding correlated with scores on episodic memory and the CDR-sum of boxes.
An exploratory whole-brain analysis in a subset of the volunteers indicated a decrease in the entorhinal cortex, though this finding disappeared upon correction for atrophy; in contrast, the hippocampal change remained robust. Van Dyck believes he will see more differences in other regions, including the association cortex, once he looks at larger sample sizes across the range of disease severity. “Our initial experience is that the medial temporal lobe changes are much bigger than anything else in early stage disease,” he said.
Going forward, van Dyck said his group will define how 11C-UCB-J relates to cognitive decline, FDG, and tau PET. They have partnered with Cognition Therapeutics Inc., Pittsburgh, to use the tracer in an upcoming trial of the anti-Aβ oligomer therapy CT1812 and see if the tracer can detect a protective effect on synapses that has been hypothesized for this drug (see Part 10 of this series). Van Dyck told Alzforum that he hopes to see data from independent groups evaluating this tracer before long.—Pat McCaffrey and Gabrielle Strobel
Automated CSF Tests: Check. Blood Tests: In the Works
As prevention trials for Alzheimer’s disease ramp up, researchers need fast and cheap ways to diagnose early, and to screen cognitively normal people. PET is expensive. Cerebrospinal fluid (CSF) biomarkers of amyloidosis and tauopathy could bring down costs, and the field is moving from manual, research-grade immunoassays to automated systems that robustly measure CSF Aβ peptides Aβ42, Aβ40, and tau within an hour’s time. At the 10th Clinical Trials on Amyloid Disease conference, held November 1–4 in Boston, several groups presented new data validating the automated tests to support their widespread clinical use. Excitingly, just as CSF tests are nearing this important finish line, the story on nascent blood tests is suddenly changing from one of failure to one of great promise. New results from two different blood tests for Aβ give a glimpse of a future with less-invasive tests speeding trial recruitment.
In the latest validation study of automated CSF assays, Chengjie Xiong, working with Anne Fagan at Washington University, St. Louis, asked if Roche’s Elecsys platform for analyzing CSF biomarkers—for which low variance continues to be established in various clinical cohorts—manages to predict clinical progression in cognitively normal older people. Xiong measured CSF Aβ42, total tau (tTau) and phosphotau (pTau) in 362 clinically normal volunteers, aged 65 or older, who had been followed for an average of five years with repeat cognitive assessments. In this time, 84 people progressed to a clinical dementia rating of 0.5 or higher. After adjusting for age, ApoE4 status, sex, and education, Xiong found that Aβ42 concentration, or the ratios of tTau/Aβ42 or pTau/Aβ42, best predicted clinical progression and cognitive decline. Concentrations of tTau or pTau alone were not associated with progression.
In a separate analysis, Fagan’s group determined cutoff values for biomarker positivity in 198 people who had undergone both lumbar punctures and PiB-PET scans. Applying the cutoffs to their new data, they found that people on the amyloid-positive side of the cutoff had twice the risk of clinical progression and faster rates of cognitive decline than those on its amyloid-negative side. The work shows that automated tests of CSF biomarkers can identify candidates for further screening and enrollment in clinical trials, the authors said.
On a second poster, Fagan and colleagues asked how well a competing automated assay, Fujirebio’s Lumipulse G Aβ42, compared with that company’s research-grade standard, the INNOTEST ELISA, at predicting brain amyloid. Fagan analyzed CSF from a cohort selected to have varying amyloid levels by PiB-PET, and used both the Aβ42 Lumipulse and the Aβ40 ELISA. Both assays gave low and acceptable variability between 3 and 8 percent. Absolute values for Lumipulse were 50 percent higher than for ELISA, but the two measures correlated well. As expected, both Lumipulse and INNOTEST Aβ42 discriminated PiB-PET-positive from -negative people better than did Aβ40. The Aβ42/40 ratio was best of all, with AUCs of 0.88 and 0.91 for the INNOTEST and Lumipulse G Aβ42 assays, respectively.
Fagan’s results broadly echo a recent clinical validation of Roche’s Elecsys assay, presented at the AD/PD conference last spring by Oskar Hansson of Lund University, Sweden (Apr 2017 conference news). In that study, cutoff values determined in the BioFinder cohort were applied to ADNI participants, in whom they successfully identified those with brain amyloid accumulation. In that study, too, the ratio of Aβ42/40 or Aβ42/tau outperformed Aβ42 alone in predicting amyloid-PET status.
A month ago, Fujirebio released its Lumipulse tTau assay, and Fagan told Alzforum that her lab is testing it currently. In the meantime, Didier Pitsi of the BARC Global Central Laboratory in Ghent, Belgium, shared his experience with both Lumipulse G Aβ42 and total tau assays in a large clinical lab. In Pitsi’s hands, Lumipulse had a 7–8 percent variance for Aβ42 and 7–11 percent for tau. That’s better than the 15–20 percent variability routinely reported for manual ELISAs in the Alzheimer’s Association Quality Control program, which sends samples to dozens of labs for parallel analysis.
Data from the limited number of quality-control runs of the automated tests available thus far are starting to show similarly consistent results. In five different labs, the Elecsys Aβ42 assay had a 5 percent variance, while Lumipulse G Aβ42 came in at 9.5 percent when compared in three labs. Floyd Sarsoza, University of California, San Diego, presented additional validation of the Lumipulse Aβ42 assay. He saw less than 3.4 percent variation on pooled CSF tested on different runs or days. The measurements remained stable over four weeks, and through three free-thaw cycles of the CSF.
Valeria Lifke, Roche Diagnostics, Penzberg, Germany, presented her company’s own validation data on the Elecsys tTau and pTau assays. They include a sensitivity of 60 pg/ml for tTau and 4 pg/ml pTau in CSF, good linearity and reproducibility, and little interference from a panel of drugs and endogenous compounds.
The results will support applications for marketing approval of the test for diagnostic use in Europe and the U.S. Roche scientists declined to answer questions about when their tests would be broadly available. So far, the Roche Elecsys Aβ42 test is approved in Europe, with the tTau and pTau assays expected in early 2018. Among Fujirebio’s Lumipulse assays, the CSF Aβ42 and tTau are approved in Europe, with Aβ40 expected soon. The tests are available for research use only in the U.S.; FDA approval will take longer.
Smelling Blood: Companies Catch on to Promise of Plasma Testing
Blood tests for Aβ and tau would enable screening of much larger numbers of potential participants for prevention trials. Like CSF tests, they likely would be cheaper than a PET scan, and many doctors and patients who demur when asked about a lumbar puncture readily agree to a blood draw. In the past, efforts to develop blood assays have foundered because Aβ concentrations in blood were 50 times lower than in CFS. Some blood cells make Aβ, and antibody-based plasma assays have been noisy. Together, this has stymied efforts to correlate blood Aβ levels with clinical status. Now, new techniques are “resurrecting blood assays,” said Fagan, and researchers are comparing blood Aβ levels directly with brain amyloid by PET.
AD in the Blood?
The baseline ratio of Aβ42/40 (TP42/40) in plasma dipped among people with MCI compared to cognitively normal folks (CN), and was lowest in those who developed dementia during a two-year follow up. [Courtesy of Virginia Perez-Grijalba, Araclon Biotech.]
For example, Virginia Perez-Grijalba, of Araclon Biotech, Zaragoza, Spain, presented data on her company’s Aβ test ELISA kit for detecting the Aβ40/Aβ42 ratio in blood. In a multicenter study carried out in Spain, France, and Italy, Perez-Grijalba and colleagues measured total plasma Aβ42 and Aβ40 in 228 people at baseline, and then again one and two years later. At the start, 83 of the group tested cognitively normal and 145 had amnestic MCI. The MCI group had a lower plasma Aβ42/40 ratio than the cognitively normal folks at all time points. After two years, 43 percent of those with aMCI had progressed to AD dementia, and their Aβ42/40 ratios dropped further. Those with a lower ratio at baseline also had lower FDG-PET and faced a 70 percent higher risk of progression to dementia over the next two years. Finally, Perez-Grijalba showed that the plasma Aβ42/40 ratio correlated with CSF Aβ42, and inversely correlated with brain amyloid levels as per PET.
The Spanish group recently published similar data evaluating their blood test in the Australian Imaging, Biomarker and Lifestyle study of aging (AIBL). In this longitudinal cohort, a low plasma Aβ42/40 ratio in cognitively normal people predicted cortical amyloid PET positivity with more than 80 percent accuracy (Fandos et al., 2017). The study has limitations: The researchers have yet to determine universal cutoff values, and there is some overlap between groups. Further studies are underway to optimize the test, Perez-Grijalba said.
Even so, her data dovetails with work by the group of Randy Bateman at Washington University in St. Louis, who measured the plasma Aβ42/40 ratio by mass spectrometry. This test is being commercially developed by C2N Diagnostics in St. Louis, a company Bateman co-founded. He found that PiB-PET-positivity came with a 15 percent lower Aβ42/40 ratio in blood (Jul 2017 conference news). At CTAD, Bateman presented follow-up data, telling the audience that the plasma test was able to assign brain amyloid status with 88 percent accuracy. “That is remarkable, because they use a different strategy to measure Aβ, and yet we see very good agreement,” said Perez-Grijalba. “We think measuring Aβ42/40 ratios in blood has good prospects as a prescreening tool in AD trials,” she concluded.
At CTAD, the advent of blood tests to reveal who has brain amyloid generated considerable buzz. Beyond Araclon and C2N, big pharma appears to have caught the whiff of opportunity. Word in the hallways had it that Roche Diagnostics has already developed automated Elecsys blood tests for Aβ42, Aβ40, and tTau assays, and is evaluating them in clinical cohorts. Richard Batrla-Utermann of Roche Diagnostics confirmed the company’s interest. “Based on the published data we are interested in this combination for identification of patients with AD pathology at a general practitioner level, to funnel them into further assessment in memory centers,” he emailed to Alzforum.
Whoever makes a good blood test first will hand the field a tool to screen large populations of cognitively normal people and speed up recruitment for prevention trials. And once an effective therapeutic is finally found, “demand for a blood test will be very high,” Bateman said.
Importantly, though, the automated CSF tests have nearly completed extensive validation studies and are nearing routine clinical use, whereas all this still awaits the nascent blood tests. They remain investigational for a while longer.—Pat McCaffrey and Gabrielle Strobel
If Jason Hassenstab has his way, an app that pings trial participants to take quick cognitive tests on their own cell phones will boost the reliability of data in future prevention studies. Hassenstab works at Washington University, St. Louis, and presented this work at the Clinical Trials on Alzheimer’s Disease conference, held November 1–4 in Boston. By prompting people to take tests several times daily over the course of a week and averaging results, the app accounts for hour-to-hour and day-to-day variation within a given person’s performance. This can greatly improve reproducibility in repeated cognitive assessments. In a separate study, Dorene Rentz of Brigham and Women’s Hospital, Boston, ran a digitized version of an old clock-drawing test through its paces. She found that the use of a digital pen revealed subtle cognitive changes that go undetected in the pen-and-paper version of the task. Importantly, scores on digital clock drawing tracked with biomarkers of amyloid and tau deposition, and neuronal metabolism.
Variability in cognitive testing is a huge problem. Its source is no mystery: During clinic visits, people may be tired from traveling, fighting a cold, or coming off a bad night’s sleep. They are anxious about being tested by someone in a white coat, and about coming face-to-face with their cognitive decline. They take back-to-back tests, in a single, long sitting. Many participants dread these extended test sessions. One year later, they return for more of the same.
With this model, replication is not assured. In fact, a “bad day” performance at visit 1 followed by a “good day” performance at visit 2 can make it seem as if the patient’s cognition improved when, in fact, it slipped.
Roller Coaster.
Day-to-day variability in testing can overshadow a true change in cognitive function. [Courtesy of Jason Hassenstab.]
For some commonly used cognitive measures, test-retest correlations—when the same person takes the test at two different times—are as low as 0.5. In the Dominantly Inherited Alzheimer Network (DIAN) Observational Study, Hassenstab showed, it can be even worse, with some young, healthy participants showing test-retest correlations in the 0.4–0.5 range. “We would never accept this low test-retest reliability in a biomarker,” Hassenstab said. To overcome this problem and still achieve statistical power, clinical trials must enroll large numbers of people. Alas, large trials are impossible in rare genetic populations, and wasteful in late-onset AD populations.
Burst testing seeks to reduce this variability by testing people briefly, for a few minutes, but repeatedly, at random times in the course of their normal daily life. A “burst” consists of seven days of consecutive testing, which is collapsed into one “visit” by averaging performance scores across the week. Burst testing can achieve test-retest correlations greater than 0.9 on some cognitive measures (Sliwinski et al., 2018). “It’s not a new idea, but our focus is on developing this method for prevention trials. I’m interested in making the data as reliable as possible,” said Hassenstab.
Grid Game.
In a rapid, mobile test of spatial working memory, the user remembers and reports where three items are on a grid, after completing an unrelated task. [Courtesy of Jason Hassenstab.]
Hassenstab developed the Ambulatory Research in Cognition (ARC) mobile app for testing cognition in preclinical AD. On their own cell phones, participants receive prompts to take a brief test four times a day for a week. In just a few minutes, they click through three tasks, which evaluate working spatial memory, processing speed, and associative memory.
At CTAD, Hassenstab first showed that scores on the phone test and pen-and-pencil tests correlated. Then he described results of testing ARC in real life in three different cohorts of cognitively normal adults. One included 10 members of the DIAN cohort, five mutation carriers and five noncarriers, whose average age was 38. In addition, he enrolled 17 older subjects from WashU’s own ADRC cohort, whose average age was 74. Both cohorts showed good compliance, completing an average of 21 to 22 sessions out of 28 per week. A community sample, recruited via social media, was a little less dutiful: The 85 participants, mostly women and with an average age of 38, completed 18 to 19 assessments per week.
With the smartphone tests, Hassenstab saw reproducibility climb as the number of sessions increased, so that after seven days, the test-retest reliability was near to, or even exceeded, his preset goal of 0.9 for each domain. Test scores varied over the seven days, but averaging the scores smoothed out a person’s ups and downs. “Our reliability is now very high. I am really happy about how this is panning out this far,” Hassenstab said.
Putting the test on personal phones, rather than providing a separate device to participants, is important to Hassenstab. “We want people to be familiar with their device when they do the testing, so it feels less like a separate task outside their daily life, and more like part of the natural use of their phone,” he explained. But bring-your-own-device has its own vagaries: Different phone models vary in important parameters, such as how quickly they display an image, and how fast they register a screen tap. iPhones are manageable, Hassenstab says, because there are but a few models, with consistent specs and operating systems. However, android phones are another story, with more than 5,000 devices whose software and specs vary widely. Inconveniently, some people change phones midway through studies.
Rhino Tap. A homemade robot makes 5,000 taps to assess the response latency of smartphones used for mobile cognitive assessments. Why put a three-D printed rhino head on the robot? "Oh, just because we could. It's for fun," said its maker. [Courtesy of Jason Hassenstab.]
Hassenstab’s group is developing rigorous validation protocols for phones. One accounts for tap latency—the time it takes for the phone’s processor to register a screen tap. Variations here can skew measures of response time, which form part of a participant's processing speed. Hassenstab’s team built a robot that taps a phone 5,000 times to determine the device’s tap latency; they then use that data as a covariate in their analysis to account for differences in phones. For iPhones, the latency is around 50 milliseconds, but for other phones the values vary a lot, Hassenstab said. When the response times that differentiate normal and preclinical AD might be just 100–200 milliseconds, knowing a phone’s lag time, and adjusting for it, matters.
Age matters. “In DIAN-TU, issues are minor because our participants are younger; they have phones and know how to use them. They easily download the app and get running,” Hassenstab said. In older cohorts, he finds more variability in people’s experience and comfort levels with the app. “This is early days of doing this with older people,” he says. “Many use smartphones, but many don’t. We find if we give them a phone, and sit with them and get the app working, mostly they are delighted to use it.”
Data Capture.
A digital pen with integrated camera and processor continuously tallies starts, stops, and stroke shapes during drawing, providing a wealth of information for analysis. [Courtesy of Dorene Rentz.]
The open-source ARC code will be freely available to anyone in the research community interested in build their own app. Hassenstab expects to release it in January 2018.
Yet another time-honored cognitive test is going digital. Rentz at Brigham and Women’s reported on DCTClock, an upgrade on the traditional pen-and-paper clock-drawing test that is being developed by Digital Cognition Technologies, a startup in Waltham, Massachusetts (Dec 2012 news).
Clock drawing is a simple and common screen for dementia. Clinicians ask people to draw the face of an analog clock showing a specific time, often 10 minutes past 11. Errors in the finished drawing spark further evaluation.
A physician looking at the paper sketch analyzes only the final product, whereas the digital version also captures the process. A digitizing pen records time and location continuously during drawing, detecting not just what people draw, but also when they hesitate, or stop and think, and how they organize the task. All told, the accompanying software analyzes 700 features of the drawing process, and compares the output to a database of more than 4,500 healthy and pathological tests to generate an objective score of cognitive function.
Big Reveal? Automated analysis detects subtle cognitive problems, even when the drawings look acceptable. The digital pen picks up deficits in drawing efficiency and information processing. It renders a composite score and flags if it’s outside the normal range. [Courtesy of Allison Byers, Digital Cognition Technologies.]
To ask if the digitized pen could pick out subtle cognitive impairment, Rentz compared scores on this test to results of traditional cognitive tests and to levels of AD biomarkers in 105 clinically normal older adults, some from the community and some in the Harvard Aging Brain Study. Everyone took a battery of traditional cognitive tests, and the 63 HABS participants also had amyloid, FDG and tau PET scans. Rentz found that digital-clock test scores correlated most closely with pen-and-paper tests of executive functioning and processing speed. People with subtle cognitive impairment, as defined by a performance more than 0.5 standard deviations below the norm on the Preclinical Alzheimer’s Cognition Composite, or PACC, scored significantly lower on the digital-clock test than people without any cognitive impairment.
Digital-clock scores also correlated with biomarkers. Worse overall performance on the test was associated with greater amyloid deposition, greater parietal glucose hypometabolism, and with a trend toward higher entorhinal and inferior temporal tau accumulation. The investigators concluded that the digital pen may offer a quick and sensitive measurement of cognitive change in preclinical AD, and of change over time in clinical trials.
The projects featured above are but two examples of a much larger number of efforts occurring simultaneously throughout the field. In them, biostatisticians are looking for how they might be able to extract a much richer data set for analysis from the cognitive test sessions researchers demand of study participants. One other example is the work of Howard Mackey at Genentech, who is collaborating with Ron Thomas at University of California, San Diego, and colleagues from the Alzheimer’s Prevention Initiative API. Mackey reported at CTAD that if AD trials were to use rate of decline as a summary measure—not change from baseline, as they currently do—then they would be able to incorporate a larger fraction of the data collected at points in between a trial’s baseline and its final visit.
A related point emphasized by numerous biostatisticians, including Mackey, WashU’s Andrew Aschenbrenner, and Suzanne Hendrix of Pentara Corporation, is that participants should be tested much more frequently than once every six months, and statistical models should be refined to optimize data from frequent measures. All of this work serves the goal of boosting the power of natural history and therapeutic studies.—Pat McCaffrey and Gabrielle Strobel
Don’t Be an Enrollment Loser: Throw Your Own Swab-a-Palooza!
The enormous challenges of recruitment loomed large at the 10th annual Clinical Trials on Amyloid Disease conference, held November 1–4 in Boston. Researchers are seeking new ways of identifying legions of eligible volunteers to enroll in clinical trials for Alzheimer’s disease. Prevention trials in particular are seeking people who are cognitively healthy or have but the subtlest symptoms. They are difficult to identify and enlist. Several speakers at CTAD cited a paper in which Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas had tallied the number of participants needed for existing clinical Alzheimer’s trials at 55,000, not even counting future prevention trials (Cummings et al., 2017). Where will trialists find the right people? “It’s all about recruitment now,” Stephen Salloway of Brown University’s Butler Hospital in Providence, Rhode Island, told Alzforum.
One place to trawl is online. Recent initiatives use web-based platforms to assemble cohorts of trial-ready volunteers who can be referred to studies quickly and en masse. Two U.S.-based online registries, the Alzheimer's Prevention Registry (APR) and the Brain Health Registry (BHR), are redefining the art of recruitment and referral. Overseas, European scientists are building their own online platform with the European Prevention of Alzheimer's Disease Consortium (EPAD). All three updated the audience at CTAD. Tools such as desktop DNA analyzers come in handy, as well.
JessicaLangbaum of the Banner Alzheimer’s Institute in Phoenix talked about GeneMatch. Part of the APR, GeneMatch supports prevention trials that enroll based on ApoE4 status. By joining the APR, the registry’s 288,000 enrollees have expressed an interest in participating in Alzheimer’s prevention studies. GeneMatch offers them ApoE genotyping via mail-in cheek swab kits (Dec 2015 news). Participants must be healthy, between 55 and 75, live in the U.S., and have never been diagnosed with MCI, AD, or dementia. A Clinical Laboratory Improvement Amendments (CLIA)-certified lab analyzes their DNA and stores the information in a secure database. GeneMatch does not disclose test results, but people may learn their status if they join a drug trial that discloses genotypes.
In the two years since it started, GeneMatch has screened more than 45,000 people, and identified just over 1,000 homozygote and 9,000 heterozygote ApoE4 carriers, Langbaum told Alzforum. The response rate is good: Two in three people who receive the swab kit mail it to the lab within 90 days.
Online enrollment as done by both APR and BHR complements local registries and other outreach efforts, but it does not replace them. “Online and local go hand in hand,” said Langbaum. For example, GeneMatch sends kits to its 25 partner sites, including one in Hawaii, where the partners distribute and collect the kits locally.
“We hold local swabbing parties, where the GeneMatch sign-up rate is 95 percent,” Pierre Tariot, co-director of the API at Banner, told Alzforum. “Jeffrey Cummings calls them Swab-a-Paloozas,” said Tariot. Weiner founded the BHR at the University of California, San Francisco.
“I spend a lot of time in my community doing outreach. People are very interested and receptive,” concurred Salloway, who hosted a swabbing party for GeneMatch recently. “At events, I ask everyone to tell five other people about what they learned, and to host swabbing parties themselves,” Salloway told Alzforum.
Does this get people into trials? Last July, on the basis of GeneMatch results, the Alzheimer's Prevention Initiative emailed 1,068 people, inviting them to screen for the Generation prevention trials program (Jul 2014 conference news). Sponsored by Novartis, the Generation 1 and 2 trials are massive undertakings, aiming to enroll 3,340 E4 carriers or homozygotes. Most of the recipients opened the email, and 493 accepted the invitation. Langbaum said she does not know yet how many ultimately enrolled in a Generation trial, but she is tracking those numbers. As of October 2017, 39 of the trial sites had received referrals in this way. “GeneMatch is Novartis’ leading recruitment tool for Generation in the U.S.,” Langbaum said.
Another way to ramp up genotype-based recruitment is the Spartan Cube, an ApoE desktop analyzer made by Ottawa-based Spartan Bioscience, Inc. A sleek, 4-inch cubed device that looks like a little aluminum speaker, it renders an ApoE genotype result within the hour. This makes it well-suited for local outreach events, where hosts can line up cubes side by side so that multiple people can learn their ApoE genotype on the spot. No mailing, no waiting.
Enrollment, Cubed.
Delivering ApoE genotyping results in one hour, Spartan’s mini DNA analyzer is a recruiting tool for prevention studies. [Courtesy of Spartan Biosciences.]
At CTAD, Sharon Cohen of the Toronto Memory Program and colleagues reported on a poster that the cube boosted standard recruitment strategies in a two-site study. For one, when the Toronto Memory Program added the cube to its annual booth offering memory screening at a “Walk for Memories” community event, uptake shot up eightfold. Many more people were interested in ApoE-cum-memory-testing than in memory testing alone, and those who learned their ApoE genotype were more inclined to then join a clinical trial. For another, when the Pacific Research Network in San Diego upgraded its weekly memory screening advertisements in a local newspaper by also offering on-site ApoE testing with the Spartan Cube, that site saw its response rate increase 4.6-fold.
However, Salloway and other investigators cautioned, this ApoE test remains research-grade at the moment because it is not CLIA-approved. This means that, at least in the U.S., Alzheimer’s clinicians will not use it to formally disclose ApoE genotype to people. Anyone who learned his or her ApoE4 genotype by way of the cube would have to repeat the test CLIA-grade as part of screening for the Generation trial, for example. Even so, Novartis is supporting the use of the Spartan Cube in Canada as an outreach tool.
Where are the Guys?
A recurring concern with online recruiting is the overabundance of women who sign up. GeneMatch participants are 78 percent female, which reflects the makeup of the larger APR, and indeed other registries and natural history studies in the field. “It’s not easy to get healthy individuals to join, and those who do are predominantly highly educated, white, and female,” Langbaum said. “That’s OK if the women are the health-info gatherers and send their men for trials, but it will be a problem if we cannot get men into studies,” she added. To do that, APR will start running ads targeting men. To address a related problem—the under-representation of African-American and Latino minorities—Langbaum said APR is updating its language and content to widen the registry’s reach, including translating the site into different languages.
So far, the registry has promoted 52 studies, Langbaum said. She wants to add more. In general, APR metrics such as open rates and click rates on its monthly newsletter exceed industry averages, indicating strong member engagement. Going forward, Langbaum will continue analyzing email subject line, content, and other parameters to figure out what works and what doesn't. “We are very interested in the science of recruitment,” she said.
APR promotes the Brain Health Registry, and several thousand APR members have joined the BHR, as well.
At CTAD, Shannon Finley, UCSF, recapped recent work at the BHR, the online longitudinal research and trial referral site launched in 2014 (Jun 2014 news). Unlike APR, the BHR asks members to provide demographic and health data, and to complete an online cognitive testing battery. Registrants are prompted every six months to repeat the cognitive testing. Like the APR, BHR refers participants to outside clinical trials.
To date, BHR has 55,500 members, and as with APR, registrants are mostly white and female. More than half are over 65, and 48 percent report memory concerns.
The BHR currently recruits for 16 clinical trials. So far, the registry has sent referral invitations to 16,000 members, of whom 4,000 responded with interest. Of those, about 2,000 have been placed into observational studies, fewer than 50 into treatment trials. Approximately 1,700 have signed up for optional data sharing between BHR and the trial site.
Perhaps the biggest challenge for the BHR is retention, Finley said. The registry has a committed cohort of more than 2,000 returnees who have faithfully completed cognitive testing every six months for 42 months. However, the six- month drop-off rate is 50 percent, Finley told Alzforum. One reason may be that BHR does not share cognitive results with its members. In a survey done this year, most respondents said they wanted to know how they did on the tests. More than half said they joined BHR to “learn more about [their] own brain health by taking cognitive tests and getting scores,” Finley said in her talk.
“Many of our registrants are the worried well—they are cognitively normal but concerned about memory. They want reassurance they are doing OK,” Finley told Alzforum. Because BHR is a research study, releasing data to participants is complicated. The tests are not diagnostic, and the investigators worry about potential harm to subjects from learning their cognitive status. Providing test results might confound future data collection. Monica Camacho, also at UCSF, told Alzforum the BHR team is working on some kind of data release, but has no timeline for implementation.
Finley emphasized that the BHR is adapting to the needs of its population and study partners in other ways, as well. For example, BHR’s original referral model involved looking at inclusion/exclusion criteria from trials and referring only prescreened BHR members to a given trial. Alas, BHR staff found that the trial sites preferred larger numbers of referrals without preselection, so BHR now offers direct-to-site referrals. In this model, BHR provides customized landing pages for trials, where people can sign up for studies directly, without enrolling in BHR. For ADNI3, for example, the BHR hosts an online recruiting site where 1,116 people so far have completed a brief series of screening questions and 296 have been provided local ADNI3 study site contact information. This model simplifies regulatory issues, too, as it does away with data sharing or online monitoring of a trial cohort through BHR.
Pairing Up. The Brain Health Registry now invites members to recruit spouses, family members, or trusted friends as study partners, as is also required by many clinical trials. [Image courtesy of BHR.]
In another new initiative, the BHR has started enrolling caregiver/study partners. AD research has established that informants such as spouses or adult children often describe early stages of decline more accurately than the affected person, who may lose insight or deny. So far, the BHR has 4,286 registered pairs, of whom approximately 80 percent are spouses and 85 percent live together. At CTAD, Rachel Nosheny of the BHR presented data from 2,000 of those dyads. She reported that, among registrants with a diagnosis of MCI or AD, study partners outperformed self-reporting at picking up cognitive decline, suggesting that informants may help identify candidates for MCI or dementia trials.
The UCSF scientists have started a co-enrollment program. This means participants in outside research studies also join BHR, which then contributes online longitudinal cognitive data. So far, BHR has co-enrolled 900 participants from three imaging studies, including ADNI and IDEAS. In addition, BHR offers Software as a Service (SaaS), which provides BHR tools for other research sites to use, without control by BHR. Sites can use a copy of the BHR, or can format a new custom-programmed interface. Two studies are currently using this service, a Parkinson’s disease study at UCSF and a dementia study in the Netherlands.
“The reason for so many different offers is that we are trying to help the field as best we can. Originally, we thought our referral model was what the field needed, but since starting, we have learned a lot. We want to meet people where they are, to get people into trials as fast as we can,” Finley told Alzforum.
Across the pond in Europe, registry building is continuing apace, as well. At CTAD, Craig Ritchie of the University of Edinburgh gave a progress update on the work of the European Prevention of Alzheimer’s Project (Aug 2016 conference news). EPAD is a large, multipronged initiative to mine observational aging studies, aka “parent cohorts,” across European countries for suitable participants in a virtual AD prevention trial registry. The participants are then characterized extensively, both on cognition and biomarkers, to form a standing cohort of deeply phenotyped people in the preclinical and earliest symptomatic stages of AD. These people can feed an envisioned platform of continuous Phase 2, proof-of-concept, adaptive secondary prevention trials.
At CTAD, Ritchie said that three years into this ambitious effort, EPAD has built the requisite components and is getting ready for the first trials to start in 2018. EPAD sites—called Trial Delivery Centres, or TDCs in EPAD-speak—started opening in May 2016. As of November 2017, around 380 participants had enrolled across 10 sites, and six more sites were due to open by the end of the year, Ritchie said.
A first look at this nascent baseline population shows that, unlike APR and BHR, EPAD thus far is enrolling men and women in equal numbers. Participants tend to be highly educated, have a family history of AD, and 85 percent have a CDR of zero, with the rest CDR 0.5. About 28 percent are amyloid-positive, especially those who are older, carry ApoE4, and have a family history, Ritchie said.
In general once a given registry cohort is in place, researchers can evaluate various screens to learn which best select trial participants from among them. Geraint Price, Imperial College London, explored ways of doing that in an older registry, the Cognitive Health in Ageing Register: Investigational, Observational and Trial. CHARIOT contains contact information for 28,000 adults between 55 and 80 without dementia, who were recruited from primary care practices in the United Kingdom (Larsen et al., 2015).
Price selected 711 registrants with subjectively reported cognitive decline who had a reliable informant and no significant comorbidities. For a brief screening pass, he used the informant AD-8 screening test (Galvin et al., 2007; Nov 2010 webinar) and the memory part of the Rey Auditory Verbal Learning Trials (RAVLT). With this combination, only 5 percent were eligible for referral for MCI trials. Next, Geraint tried the full RAVLT, the Montreal Cognitive Assessment, and the Informant Questionnaire on Cognitive Decline (IDCODE). That combination sent more participants into an expert adjudication process and increased referral eligibility to 19 percent. Such data suggests that brief prescreening tools including a delayed recall/recognition component, along with a clinical adjudication procedure, can increase the screening sensitivity for MCI, while self-reported cognitive difficulties do not.
What about mining electronic health records, aka EHR? After all, they hold a wealth of information on millions of people. Could they offer recruitment opportunities? Unfortunately, no, said Peter Schueler of the clinical development research organization ICON in Langen, Germany. Schueler compared the BHR population and two clinical-data aggregation providers, TriNetX in the U.S. and InSite in the European Union. Together, the clinical data covered nearly 50 million people, 135,000 of whom were diagnosed with AD. However, the EHR populations were far sicker than Brain Health participants, with high levels of heart disease, hypertension, diabetes, cancer, and stroke. In addition, both quality and completeness of the data in the EHRs fell short of what is needed to prescreen for trials, Schueler found. Only registries and study-sponsored web pages can pre-identify trial candidates, he concluded.
These entities need to boost incentives for people to participate, by providing feedback on ongoing testing or enabling members to interact and share data with other members of a given online community. As a success story, Schueler cited PatientsLikeMe, which has grown to 600,000 members and at present attracts about 10,000 new sign-ups per month. “We need a patient-centered ecosystem of AD-specific EHRs, where the participant owns their data and puts them in the registry. It would be a moving, living system. The tech is in place to do that, we just need to establish it,” Schueler said.—Pat McCaffrey and Gabrielle Strobel.
Verubecestat Negative Trial Data: What Does it Mean for BACE Inhibition?
The room was packed at the recent Clinical Trials on Alzheimer’s Disease conference, when Mike Egan of Merck & Co. presented much-anticipated data of EPOCH, the first efficacy trial of his company’s BACE inhibitor verubecestat. An expert panel stood ready to field audience questions, and discussion spilled into the hallways, receptions, and dinner conversations.
Merck had terminated the trial nine months before (Feb 2017 news), so no one expected a positive outcome. Still, researchers were anxious to hear details, and they were particularly hoping to see a sign—any sign—that verubecestat might have shown at least a slight trend toward a benefit in the more mildly symptomatic subgroup of this mild to moderate AD trial. That would have been encouraging news to other companies that are currently enrolling for large, expensive trials of their own BACE inhibitors in essentially the same band of the Alzheimer’s disease continuum as the milder half of patients in this trial by Merck.
Alas, it was not to be. Egan said verubecestat showed no clinical benefit whatsoever in moderate or mild AD. If anything, a subgroup analyses Egan presented suggested a trend for worsening on verubecestat. The drug’s side effect profile in this trial was good enough to continue evaluating verubecestat in earlier-stage trials, though it did include psychiatric findings.
To some experts, the overall data simply meant that verubecestat is ineffective at this stage of AD but may work earlier. To them, it said nothing about the prospects of other BACE inhibitors. “This was a milestone trial. Merck did a convincing study to show this drug does not help in mild to moderate AD. It is one trial in one population,” said Jeff Cummings of the Lou Ruvo Center for Brain Health in Las Vegas. “I have every confidence that verubecestat, or another BACE inhibitor, will be successful in a prevention paradigm,” concurred Paul Aisen of USC’s Alzheimer’s Therapeutic Research Institute, San Diego.
To other experts, the data implied that perhaps verubecestat did have a cognitive effect that was concealed by a second, neuropsychiatric effect. Yet others argued that a BACE inhibitor ought to be paired with a plaque-removing antibody to prevent leakage of Aβ oligomers from plaques. Commentators applauded that Merck simply laid out the data, warts and all, without trying to massage it or read hopeful interpretations into underpowered post hoc analyses.
Egan noted that because verubecestat previously had been evaluated only for safety and target engagement, and skipped a conventional Phase 2, the Phase 3 part of the present EPOCH Phase 2/3 trial delivered the first set of efficacy data on this compound. Phase 3 randomized 1,958 people with AD, whose MMSE was between 15 and 28, to receive either 12 mg or 40 mg of drug or placebo for 18 months. Its participants included slightly more women than men, 80 percent Caucasians, and 63 percent with at least one ApoE4 allele. Most took a cholinesterase inhibitor and/or memantine. The trial engaged 238 sites in 21 countries in Europe, North America, and Japan.
Verubecestat was evaluated on a co-primary outcome of ADCD-ADL and ADAS-cog11. On both these measures, the curves of placebo and both verubecestat doses were almost superimposed—there was no statistically significant difference between the groups at any time point. If anything, on the ADAS-cog11, both verubecestat dose groups appeared to edge away ever so slightly from the placebo curve in the direction of worsening cognition at all but one time point after baseline. Egan also shared prespecified subgroup analyses of the ADAS-cog outcome by ApoE genotype and disease severity. This data hinted that ApoE4 noncarriers may have had a slight dose-dependent response in favor of placebo, not the BACE inhibitor. “It was very sad for all of us to see these results,” Egan told Alzforum.
One secondary outcome was the CDR sum of boxes. It showed no change between the treatment arms, indicating dementia worsened similarly in all groups. Another was total hippocampal volume, which shrank slightly faster on treatment than placebo. In response to an audience question, Egan said that whole-brain and ventricular volume showed similar small but treatment-related changes. This MRI finding echoes that of previous trials going back to the beginning of anti-amyloid therapy (Jul 2004 news), but the reason remains unclear. It could be related to reduced inflammation or plaque, Egan said.
The trial’s small flutemetamol PET sub-study indicated a 2 percent amyloid plaque reduction on the verubecestat low dose and a 4 percent reduction on the high dose. “Though these changes are small, they do suggest that verubecestat engaged its target in the brain,” Egan said. As expected from verubecestat Phase 1 data, the CSF sub-study of EPOCH showed a 70 percent reduction of Aβ40 on the low dose, and nearly 80 percent reduction on the high dose. Other scientists agreed that this data constitutes evidence of target engagement.
How safe and tolerable was verubecestat in these patients? Adverse events were significantly more frequent on drug than placebo, but not in the way the field might have expected. For example, Merck closely monitored vision and skin discoloration, in part because mouse studies had raised flags. Nothing untoward happened on these counts, Egan showed.
There were other notable side effects, however. Dropouts due to side effects were significantly more common on the high dose than low dose or placebo. Among the prespecified side effects, neither ARIA nor delirium was a problem; however, 3.5 times as many people on drug than on placebo developed rashes, which were treatable, Egan said. More worrisome to some observers were psychiatric effects such as anxiety, suicidal ideation, and insomnia, as well as falls and weight loss.
Eric Siemers of Eli Lilly and Co. asked if the totality of these side effects could perhaps mean that verubecestat has multiple effects in the brain, where one worsens the patient’s neuropsychiatric status and in this way possibly masks a parallel, cognitive effect. This point echoed in hallway discussions. Some prominent clinicians expressed concern about the psychiatric effects, while others said that in older people with symptomatic Alzheimer’s, the brain can be so ill and fragile that such effects are common with any centrally active drug, even memantine. “It would be better if we had not seen those effects, but I would not expect them to show up in younger, cognitively normal people,” said Randy Bateman of Washington University in St. Louis. Bateman is in the process of selecting a BACE inhibitor for evaluation in a DIAN-TU primary prevention trial of autosomal-dominant mutation carriers.
Lawrence Honig of Columbia University Medical School in New York articulated an unspoken question in the wake of a major trial showing that target engagement yielded no clinical benefit. “The big concern the field has now is whether this a class effect or a molecule effect,” Honig said. He hopes that the results will not come to resemble those of semagacestat and avagacestat, γ-secretase inhibitors that proved deleterious to cognition. Will this data slow enrollment of other BACE inhibitor trials? “I don’t think so. We need to do the experiment. There is enormous need for advances in AD treatment, and hope usually trumps nihilism,” Honig told Alzforum.
Tobias Bittner of Roche asked why a BACE inhibitor would reduce the PET amyloid signal. After all, the drug itself does nothing to plaques, but it could affect the balance of Aβ production and clearance. Speaking on background, other scientists speculated that verubecestat, by stopping monomer production, could have shifted an equilibrium of monomer versus oligomer/protofibrillar Aβ species to the left. That could have allowed oligomers to leach out of plaques, where they could have affected nearby synapses and impaired their function. This could explain the trial’s overall finding that Aβ monomer was dramatically reduced and amyloid plaques slightly reduced, yet cognition slightly favored placebo, if anything.
If that were the case, several leading scientists agreed, then BACE inhibition in people with significant amyloid load might have to be paired with plaque removal. The idea of combining a BACE inhibitor and an antibody to both shut off production and remove plaques so that smaller species will not leach out appealed to many scientists, and renewed calls to move forward with combination trials were heard throughout CTAD.
On the other hand, the antibody solanezumab reduced monomeric Aβ and did achieve a small, if insufficient, cognitive benefit in mild AD. Scientists at CTAD were puzzled by the comparison.
To Aisen and others, the main point of the data available thus far was that verubecestat is an oral drug that is safe, well-tolerated, engages its target in the brain, and stands a better chance of success in prevention trials. “Mild to moderate dementia is end-stage disease with a heavy fibrillary amyloid load, extensive tau pathology, and neurodegeneration. BACE inhibition is going to belong to earlier stages of the disease process,” Aisen said. A Phase 3 trial of verubecestat, aka MK-8931, in prodromal AD is ongoing.
Above all, many scientists at CTAD emphasized that negative trials are important. “Negative studies occur in most fields, and are necessary for learning,” said Maria Carrillo of the Alzheimer’s Association. She added, “We are trying to treat and prevent dementia, one of the most difficult things science is doing in this century. We have to work together and share negative and positive findings.” Many others agreed. “We can only advance the field if we do this kind of trial,” Cummings said.—Gabrielle Strobel
At this year’s Clinical Trials on Amyloid Disease meeting, held November 1–4 in Boston, Roche scientists showed that high doses of gantenerumab reduced amyloid plaques in the brains of people with Alzheimer’s disease, and that raising the dose slowly mitigated the risk of side effects. Results from the ongoing open-label extensions (OLE) of two Phase 3 trials indicated that monthly injections under the skin of 900 mg and 1,200 mg gantenerumab reduced brain amyloid by up to 15 percent over six to nine months. For one-third of treated patients, uptake of the amyloid PET ligand florbetapir dropped below a predetermined threshold for positivity. Researchers at the meeting said the data were encouraging, though they cautioned that the number of patients was small and interpretation tricky because participants received different doses of drug for different periods of time.
“I am highly optimistic about the late-onset sporadic AD data from the OLE PET study, which clearly demonstrated a biologically significant decrease in amyloid over six to nine months. This demonstrates that one of the main pathologies of AD can be reversed with gantenerumab,” said Randy Bateman of Washington University, St. Louis.
The various dosing regimens came from a need to reduce the risk of amyloid-related imaging abnormalities (ARIA), namely vasogenic edema (ARIA-E) and micro-hemorrhages (ARIA-H). The former in particular is a common side effect of amyloid immunotherapy. At CTAD, Rachelle Doody, who left Baylor to join F. Hoffmann-La Roche, Basel, Switzerland, outlined the rationale for the OLE protocols for the Scarlet RoAD and Marguerite RoAD trials, neither of which had shown efficacy on primary endpoints (Dec 2014 news). Pharmacokinetic and pharmacodynamic analysis from the halted Scarlet RoAD trial suggested that, even though the trial was negative on the primary endpoint, participants who had had the highest gantenerumab exposure, and those in whom disease progressed fast, did appear to benefit both in amyloid reduction and ADAS-cog, respectively (Nov 2015 conference news).
Amyloid Cleared? Five patients titrated up to high-dose gantenerumab saw a reduction in florbetapir SUVR of up to 0.75. [Courtesy of Gregory Klein, F. Hoffmann-La Roche.]
A model built with data from Scarlet RoAD and with published data on another therapeutic antibody, Biogen’s aducanumab, predicted that amyloid would need to be reduced in the brain by at least 20 percent to see a slowing of cognitive decline, said Doody. The 105 mg and 225 mg doses used in Scarlet RoAD would not cut it. Instead, the model predicted that 1,200 mg of gantenerumab subcutaneously would be needed over a two-year period. Indeed, the DIAN-TU trial of gantenerumab in people with autosomal-dominant AD mutations increased the dose subsequent to this analysis.
The problem at high doses would be ARIA. Roche scientists then modeled gantenerumab’s likely effect on ARIA, taking into account dose, duration of treatment, and ApoE genotype—a known risk for ARIA. That model informed the dosing regimen in the open-label extension studies. Patients who wished to continue treatment would eventually receive the 1,200 mg dose, but would be titrated up based on their prior exposure to the drug and their ApoE4 status. Hence, patients started on either 105, 225, 300, 450, or 600 mg doses, and got higher doses every two months until they had reached 1,200 mg. Some patients ended up on the high dose for 10 months, others for only two, all while being monitored for ARIA by way of MRI scans. If patients experienced significant ARIA, they paused treatment until it resolved.
Doody said that the titration reduced ARIA-E as expected. The model predicted that most ARIA-E would occur during the first few months, then taper off even as the dose went up further, and that is what happened. At CTAD, Doody showed closely overlapping plots of predicted versus recorded ARIA-E over time.
Mirjana Andjelkovic, also from Roche, offered details in a talk and a poster. Out of 228 patients who enrolled in the Marguerite RoAD OLE, 203 had at least one MRI post-baseline and 58 of them had ARIA-E. For most, it was asymptomatic. Fourteen people had a mild headache or confusion; three developed more serious symptoms, two having a seizure that resolved, and one a possible stroke.
Roche used two titration protocols—one reaching the target dose of 1,200 after six months, the other after 10 months. Because Scarlet RoAD participants were taken off gantenerumab for more than a year before the open-label extension started, their Alzheimer’s had progressed. Even so, Andjelkovic said, overall safety findings were unchanged on the higher dose.
Patients who were titrated up more slowly had less ARIA-E. For example, among ApoE4 carriers who eventually received 1,200 mg, 16 percent developed ARIA-E if started on 225 mg as compared to 30 percent of those who started on 400 mg. Similarly, the incidence among ApoE4 noncarriers who started on 600 mg was double that among those starting on 300 mg.
Of the patients who experienced ARIA-E, 35 also had signs of ARIA-H, as did 15 patients who tested negative for ARIA-E. Clinicians generally consider ARIA-H less of a concern, and Doody noted that it crops up in placebo groups as well. In summary, Doody felt that higher doses of gantenerumab could be used safely. “I think it is possible we can optimize the schedule to reduce ARIA and to use the same titration for both ApoE4 carriers and noncarriers, which would make trials more manageable,” Doody said.
What about amyloid reduction? At CTAD, Gregory Klein, also from Roche, showed the first data from an 81-patient PET sub-study of the OLE trials. Klein focused on patients deemed to have sufficient antibody that it might move amyloid from the brain, i.e., people who had received at least six doses of 900 mg or 1,200 mg during the OLE period. Forty patients fit the bill so far. Klein subdivided them into three bins: nine patients who had continued from Scarlet RoAD; 14 from the original Marguerite RoAD placebo group; and 17 who had been in that trial’s treatment group. The distinctions matter because these patients started out with different levels of amyloid and had different exposures to high-dose gantenerumab in the OLEs. Scarlet RoAD enrolled people with prodromal AD, whose baseline florbetapir standard-uptake value ratio (SUVR) in the OLE rang in at 1.45. Marguerite RoAD enrolled people with mild AD, which was reflected in higher baseline OLE SUVRs of 1.79 and 1.7, for those in the original control and treatment groups, respectively. Further, Scarlet RoAD participants had, on average, 5.7 months of high-dose immunotherapy in the OLE, whereas for those from the Marguerite RoAD control and treatment arms it was 6.8 and 9.4 months.
Klein showed that SUVRs for the nine Scarlet RoAD patients fell by 0.13, or 9 percent. The drop doubled in the Marguerite RoAD groups to 0.24 and 0.27, or about 15 percent. Klein said this fit with Roche’s model, which had predicted a 26 percent reduction in amyloid over 20 months of high-dose treatment. For one-third of the patients the SUVR had dropped below the predetermined cutoff for amyloid positivity. At 1.4, the cutoff is higher than typically used in the field. It was determined ad hoc by comparing uptake from six cortical regions and normalizing to the cerebellum. Klein noted that absolute SUVRs are not comparable between methodologies.
Researchers at CTAD found Klein’s data promising, but asked about effects on cognition. Do people with more amyloid removal decline less? Klein replied that because it has no placebo group and few participants, the OLE is not powered to answer this question, though as participants are continuing on treatment they are being assessed for cognition.
Others wondered what might happen when treatment ends. Will brain amyloid return? Researchers are studying this question. “We will have two-year scan data, and we are conducting retrospective analyses of earlier studies to see what happens when dosing stops,” Klein said.
Roche is planning two new Phase 3 clinical trials, called GRADUATE 1 and 2. According to a company spokesperson, starting in early 2018 each trial will enroll 750 people with prodromal to mild AD, and treat them for two years with subcutaneous injections of antibody or placebo. Roche did not disclose the dose except to say it will be higher than in the previous trials, and the trials will employ a single titration scheme for both ApoE4 carriers and noncarriers.
The gantenerumab findings are in sync with CTAD presentations on the anti-Aβ antibody aducanumab. Biogen researchers showed marked amyloid reduction on the highest dose with long-term exposure, and mitigation of ARIA with a dose titration protocol (see Part 1 of this series).—Tom Fagan
Pimavanserin Trial Raises Hope for Treating Dementia-Related Psychosis
Advancing dementia can bring with it psychosis, which greatly adds to the burden of life with Alzheimer’s disease for both the sufferer and his or her carers. No drugs are approved to treat dementia-related psychosis. Used off-label, psychiatric drugs tend to offer little benefit but much risk, from faster cognitive decline to higher mortality due to stroke and other causes.
The serotonin 5-HT2A receptor antagonist pimavanserin was recently approved for treating psychosis related to Parkinson’s disease, and at the Clinical Trials on Alzheimer’s Disease conference, held November 1–4 in Boston, Clive Ballard of the University of Exeter Medical School in the U.K. told the assembled scientists that pimavanserin now looks promising for Alzheimer’s, too. In a Phase 2 study, a six-week course of pimavanserin significantly reduced psychosis in people with AD, with no significant adverse effects and no negative effect on cognitive function.
“This is a beneficial treatment. We have to do a Phase 3, but compared to very high adverse event rates with previous antipsychotics, this seems like a large step forward,” said Ballard.
Pierre Tariot, Banner Health Institute, Phoenix, concurred. “We use current antipsychotics at our peril. With them, the people who benefit without being harmed are few. I hope pimavanserin will improve this situation,” Tariot told the audience.
Developed by Acadia Pharmaceuticals, San Diego, California, pimavanserin won FDA approval in 2016 for the treatment of hallucinations and delusions associated with Parkinson’s disease (Apr 2016 news).
To test the drug in psychosis related to dementia, Ballard’s trial treated 181 Alzheimer’s patients from 133 nursing homes in the U.K. with 34 mg of pimavanserin or placebo once daily for six weeks, with an additional six-week extension. Participants were mostly women in their mid- to late 80s, with possible or probable AD and psychotic symptoms that developed after dementia started. One-third had severe psychosis, and all had at least mild symptoms every day, or severe symptoms once a week or more.
The trial’s primary endpoint was the NPI-NH psychosis score at six weeks, and secondary endpoints measured global cognition, agitation, and safety. Ballard said he included broad assessments such the ADCS-Clinical Global Impression of Change to measure general functioning that might also flag any safety issues. The analysis included patients who took at least one dose of medication and had at least one post-dose psychiatric assessment.
Blue is Better. Responder analysis indicates how many patients improve, and by how much, in a clinical trial. It is a measure of clinical relevance. More people on pimavanserin (blue) bettered their psychosis scores than those on placebo (gray). [Courtesy of Clive Ballard.]
Over the six-week treatment, psychosis scores declined in both pimavanserin and placebo groups, but more so in the former. This group experienced nearly 4 points of reduction in the NPI-NH score, where lower scores are better. This was 39.5 percent more than seen in the placebo group. Just over 55 percent of the participants showed a response to drug (defined as a 30 percent improvement in score), compared to 37 percent responding to placebo. Both the drop in NPI-NH score and increased response rate were statistically significant. This effect size exceeded that of previous trials of atypical antipsychotics, Ballard said.
He saw no relapse when the drug was continued for another six weeks; however, the placebo group continued to improve, such that there was no longer a difference between placebo and treated groups by 12 weeks. “The benefit is substantial, but we cannot say if it was sustained,” he said.
Stephen Salloway of Brown University, Providence, Rhode Island, questioned this timing, where the treated group did better than placebo at six but not 12 weeks. Ballard noted that the decline in NPI-NH scores in the placebo group over 12 weeks was consistent with the known remitting-recurring nature of psychotic symptoms in people with AD. Indeed, 70 percent of patients improve over 12 weeks, though half will relapse within a year. “Either pimavanserin has an acute effect that is lost later, or the symptoms are remitting-recurring,” Ballard said. “We need to do a longer trial to see if pimavanserin can prevent their recurrence.”
Worse Symptoms, More Relief. The majority of people with severe psychosis responded to pimavanserin (blue). Nearly 80 percent of patients showed greater than 50 percent improvement after six weeks, twice that seen in the placebo group (gray). [Courtesy of Clive Ballard.]
Patients whose symptoms were worse at baseline showed a better response, and pimavanserin reduced psychosis symptoms by 66 percent in people with severe psychosis. People who had used antipsychotics before, or whose baseline scores for agitation/aggression were high, also responded well to pimavanserin. People with concurrent severe psychosis and agitation had the greatest benefit. “Anything that’s indicating higher severity predicts a better response,” Ballard said. There was some suggestion of greater benefit with concurrent SSRI use.
The treatment raised no new safety flags over previous trials. The drug group did have more serious adverse events compared to placebo, but few people discontinued the drug. In particular, the investigators saw no increase in falls, fractures, stroke, or mortality—all concerns with atypical antipsychotics. The one new safety sign was that a small proportion of the drug-treated group lost weight, which had not been seen in previous trials. This needs to be studied further, Ballard and others at CTAD agreed.
Importantly, preliminary results suggested that treatment did not affect cognitive function, as the MMSE stayed stable in both groups over the 12-week trial.
In September, Acadia Pharmaceuticals began a six-month Phase 3 trial to evaluate pimavanserin’s ability to prevent relapse of psychosis in patients with Alzheimer’s or other dementias, including dementia with Lewy bodies, Parkinson’s disease dementia, and vascular and frontotemporal dementia. Participants whose symptoms stablilze after 12 weeks on pimavanserin will be radomized to drug or placebo, and then followed until relapse. Acadia is also running a 1-year open-label Phase 2 trial of pimavanserin in people whose AD is accompanied by agitation and aggression.
The need for new medications in dementia has only grown as nursing homes have begun to rein in excessive use of antipsychotics in these patients (Gurwitz et al., 2017).—Pat McCaffrey.
Blood, the Secret Sauce? Focus on Plasma Promises AD Treatment
In mice, infusing young blood rejuvenates the old, and even staves off some of the changes linked to Alzheimer’s. It’s too early to say if the same is true in people, but first results look encouraging. At the 2017 Clinical Trials on Alzheimer’s Disease meeting, held November 1–4 in Boston, Sharon Sha of Stanford University in Palo Alto, California, presented results of a small Phase 1 trial in which patients with mild to moderate AD received infusions of plasma donated by young men. The treatment appeared safe, and gave recipients a small boost on tests of everyday functioning. In a press release, Karoly Nikolich, from the trial sponsor, Alkahest, Inc., San Carlos, California, wrote that the data support “the ability of plasma compositions to counteract the biological processes underlying neurodegeneration.” The company has since made a proprietary plasma fraction to be its lead clinical candidate, and will evaluate it as a potential treatment for mild to moderate Alzheimer’s disease, Nikolich wrote.
In another plasma-based approach, Montserrat Costa, who works at the blood products company Grifols in Barcelona, Spain, presented data from that company’s ongoing testing of repeated plasma exchange and albumin replacement to treat symptoms of AD. The idea is that albumin’s propensity to bind Aβ creates a peripheral “sink” that coaxes the toxic protein from the brain, though results from ongoing trials suggest the treatment could also augment antioxidant defenses.
The Alkahest trial was inspired by work in Tony Wyss-Coray’s lab at Stanford. Back in 2011, Wyss-Coray and colleagues demonstrated that parabiotic joining of the blood supplies of a young and an old mouse reversed the effects of aging on the brain (Aug 2011 news). Subsequently, the researchers bypassed the need to conjoin the animals and succeeded in boosting neurogenesis, synaptic activity, and memory in old mice by simply injecting young plasma (May 2014 conference news). Young plasma even improved memory in mouse models of AD (Sep 2016 news).
Deep Cleanse.
Plasmapheresis equipment in a treatment room at Fundació ACE in Barcelona, the site of a trial of complete plasma exchange and albumin replacement in people with AD. [Courtesy of Fundació ACE.]
Intravenous administration of plasma—the soluble portion of blood sans its cells—is routine during surgery and trauma, and as a treatment for clotting factor deficiencies. In the Plasma for Alzheimer Symptom Amelioration study (PLASMA), Sha assessed the safety and feasibility of plasma infusions in people with AD.
For the trial, Sha enrolled 18 patients with mild to moderate AD, with an average age in the early 70s and average MMSE score around 19. For the first nine patients, she used a double-blind, placebo-controlled crossover design: After baseline cognitive testing, patients received four weekly infusions of either one unit, i.e. 250 ml, of plasma from men betweem the ages of 18 and 30, or one bag of saline. Then all cognitive tests were repeated. After a six-week washout period, the patients repeated the same sequence but received the opposite treatment from the first block. Of the nine who went through the first round, two left the trial before beginning the second round of treatment. Sensing study fatigue, the investigators switched to open-label, treatment-only infusion for the remaining nine subjects. In total, 13 completed active treatment and nine received placebo.
The treatment appeared safe, with no serious adverse events associated with plasma infusion. One person dropped out due to hives, and one due to an unrelated stroke.
In treated participants, Sha saw a statistically significant change in function, with a 4.5-point improvement on the Functional Activities Questionnaire and an increase of 3.2 points on the ADCS-Activities of Daily Living over those who got placebo. There was no improvement in the CDR-SB, a combined clinical-functional endpoint. Sha also detected no improvement in overall cognition measured by the ADAS-Cog or Trails test. Some domains of the ADAS-Cog shifted slightly: Commands were improved, while word recognition and comprehension declined. Participants had no changes in mood assessed by two methods.
Did the improvement reflect a placebo effect? Sha does not think so. Individual patients’ scores indicated that four of six participants treated in the crossover (blinded) arm improved on the ADCS-ADL, while only two of the seven open-label patients did. This makes it unlikely that responders in the open-label arm drove the improvement in average scores, she said, though others noted that the numbers are so small as to render the results vulnerable to chance.
Anecdotally, Sha told Alzforum that study partners commented that their participant seemed “brighter and more engaged” after treatment, and there was a trend for improvement on memory. “Some study partners planned outings and museum visits for the days after a treatment visit, because they expected dad will be more lucid and energetic,” Nikolich told Alzforum. The results warrant exploration in a larger, longer study, the scientists concluded
Future efforts will focus on a proprietary plasma fraction of some 400 proteins that Alkahest is developing, rather than whole plasma, Sha said. The fraction is easier to use, as it requires no matching of blood type. To produce this fraction in large batches, Alkahest is partnering with Grifols, one of the world’s largest producers of plasma products, which invested US$50 million in the company in 2015.
Separately, Grifols is also working on its own plasma-based therapy. Called PE-A, the treatment comprises a total plasma exchange and albumin replacement. Albumin, one of the body’s most abundant proteins, binds 90 percent of circulating Aβ, and provides the predominant antioxidant activity in plasma. PE-A aims to remove Aβ-laden, oxidized albumin and replace it with fresh protein, thereby lowering amyloid levels, offering binding capacity for additional Aβ, and boosting protection against oxidative stress.
The idea was put to the test in a placebo-controlled Phase 2 trial that treated 42 patients with 18 plasma exchanges over the course of six months (Boada et al., 2017). The exchanges decreased plasma Aβ42, and caused a borderline increase in CSF Aβ42 at the end of treatment that reversed after treatment stopped. Two cognitive measures, the Boston naming test and a semantic verbal-fluency test, improved from baseline in treated patients, and the ADAS-Cog and MMSE showed a trend in the right direction.
At CTAD, Costa presented data on the characterization of albumin from patients in the trial, looking at the state of oxidation and other post-translational modifications. She found that plasma or CSF albumin was more oxidized at baseline in AD patients compared to healthy controls, suggesting it was less able to buffer oxidative stress. The difference in CSF was large and consistent enough that it clearly distinguished patients from controls.
In the Phase 2 study, Costa found no significant change in albumin’s oxidation status after treatment. However, treatment did cause a reduction in glycated albumin, a sugar-modified form of the protein that has been reported to have toxic properties (Ramos-Fernandez et al., 2014).
Overall, there was a trend toward higher plasma levels of native, unmodified albumin at the end of treatment, at the same time point when CSF Aβ42 was slightly higher. Costa suggested that if this relationship can be confirmed in a larger cohort, albumin modification could serve as a biomarker, and possibly a therapeutic target for treatment. Grifols is now running a randomized, multicenter Phase 2b/3 trial in nearly 500 patients. It will finish in 2018, hopefully with a definitive answer, Costa said.—Pat McCaffrey and Gabrielle Strobel
Elusive or Not, Aβ Oligomers Are in BioPharma Crosshairs
They are difficult to isolate, hard to quantify, and virtually uncharacterized structurally. No matter the maddening vagueness of the target, scientists think they have therapies to go after Aβ oligomers. At this year’s Clinical Trials in Alzheimer’s Disease meeting, held November 1–4 in Boston, researchers outlined a handful of strategies to sweep these toxic molecules out of the brain. They include small molecules said to prevent oligomer formation or block their toxicity, such as the sigma-2 antagonist CT1812 and the glutaminyl cyclase inhibitor PQ912. They include immunotherapies, both known ones such as crenezumab and two newcomers called KHK6640 and UB-311. Even the failed-and-resurrected tramiprosate is being presented to fit the bill. Most of these drugs are in early clinical development.
CT1812 Lon Schneider, University of Southern California, Los Angeles, reviewed safety data from a multicenter, double-blind, placebo-controlled Phase 1b/2a study of CT1812, which was developed by Cognition Therapeutics, Inc., in Pittsburgh. Susan Catalano and colleagues at that company identified this molecule as a sigma-2 receptor allosteric antagonist that prevents Aβ oligomers from binding oligomer receptors and compromising synaptic function (Dec 2014 conference news). A prior Phase 1 trial conducted in Australia indicated that the compound was safe and well tolerated up to a dose of 560mg in healthy elderly volunteers.
The second Phase 1b/2a trial enrolled 19 patients with mild to moderate AD between the ages of 50 and 80, whose MMSEs ranged from 18–26. They were randomized to receive 90, 280, or 560 mg CT1812 or placebo once daily for 28 days. While incidence of treatment-emergent adverse events was the primary outcome, the study also measured cognition, a fleet of CSF biomarkers, and changes in protein expression patterns in the CSF and plasma.
Schneider reported that most patients, including 60 percent of placebo-treated patients, experienced mild, transient, unintended effects, including nausea, vomiting, headache, post-lumbar puncture headache, fatigue, and lethargy. The incidence of adverse events was slightly higher in the highest dose group, he said. Four patients developed a reduction in T cells in the blood called lymphocytopenia, which resolved before the end of the study. “Generally, the drug is safe and well tolerated at the doses given,” said Schneider.
The study was not powered to detect changes in cognition, and clinicians observed no change on the ADAS-Cog14, a verbal-fluency test, or a category-fluency test.
Both Schneider and Catalano claimed that the trial yielded evidence for synapse protection. Collaborators at Kaj Blennow’s lab at the University of Gothenburg, Sweden, used a sandwich ELISA to measure fragments of neurogranin in CSF samples from the trial. Considered a marker of postsynaptic damage, neurogranin fragments tick up in AD CSF compared to controls (Jan 2015 news; Kvartsberg et al., 2014). Indeed, over this 28-day trial, CSF neurogranin rose slightly in the placebo and fell slightly in the treated groups. There was no apparent dose response, however. The difference from placebo was statistically significant only in the 90 mg group and pooled treatments groups, which had 33 and 18 percent less neurogranin, respectively.
More indirect evidence for neuroprotection came from liquid chromatography–mass spec analysis of CSF and plasma samples carried out by researchers at Caprion Biosciences, Inc., in Montreal. Catalano reported that synaptotagmin-1, another synaptic marker that is elevated in AD CSF (Öhrfelt et al., 2016), decreased 59 percent in treated patients compared with controls. Catalano told Alzforum that changes in expression of additional proteins supported the idea of a synaptic benefit, as well. Cognition Therapeutics has filed a patent on a protein signature she considers a promising indicator of target engagement; she expects to disclose the signature next year. “We need to replicate these findings in another, longer-duration clinical trial and look at them in the context of cognitive function,” she said.
Four new trials for CT1812 are planned. One, in collaboration with Yvette Sheline at the University of Pennsylvania, Philadelphia, and John Cirrito at Washington University, St. Louis, will examine CSF hourly after single injections of CT1812 to look for displacement of Aβ oligomers from the brain parenchyma. In another, Philip Scheltens and colleagues at VU Medical Center, Amsterdam, will use EEG to look for evidence of synaptic network changes in a one-month placebo/drug crossover design trial. With the same goal in mind, Chris van Dyck and Richard Carson and colleagues at Yale University, New Haven, Connecticut, will use the investigational PET tracer UCB-J in a third trial to look for changes in synaptic vesicle glycoprotein 2A, a marker of synapse count, (see Part 2 of this series; Jul 2016 news). This six-month, NIA-supported trial will also conduct FDG PET and fMRI scans on, and collect CSF from, 21 volunteers taking placebo or one of two drug doses. Catalano said participants may opt to donate brain tissue after death to enable a comparison between in vivo UCB-J binding and postmortem counting of synapses. The fourth trial, a Phase 2 of 160 patients, will be similar in concept, but use less imaging, Catalano told Alzforum.
For another early stage treatment, the glutaminyl cyclase (QC) inhibitor PQ912, Scheltens reviewed previously presented data from the Phase 2a SAPHIR trial. The QC enzyme cyclizes the leading glutamate in N-terminally truncated Aβ3-x peptides, turning them into pyroglutamate forms (pGlu-Aβ), which are highly prone to oligomerize and aggregate (Apr 2008 conference news; Mar 2013 conference news). The drug appeared largely safe, though treated participants had more gastrointestinal problems and skin reactions than those on placebo. CSF analysis indicated a 91 percent inhibition of QC, and a future trial will focus on dose-ranging to find a more tolerable but still-effective dose.
In this trial, the median CSF GluAβ oligomer level in the treatment group dropped from 0.51 arbitrary units to 0.44 AUs by the end of the trial. The study used a proprietary assay to quantify this species of Aβ. By comparison, the median pGluAβ oligomer level rose from 0.01 AU to 0.26 AU in the placebo group. Overall pGluAβ oligomer fell by 18 percent in treated patients while increasing by 17 percent in controls. Rachelle Doody of F. Hoffmann-La Roche in Basel, Switzerland, wondered why the baseline values were so different between controls and treatment groups. Scheltens said this likely reflected difficulty in measuring oligomers. “The assay is not that sensitive, so we have to be very careful in how we interpret the data,” he said. “The only message I want to convey is that upon treatment over time, the amount of measurable oligomers went down in the active group and up in the placebo group, suggesting an effect of the drug on these oligomers.” He noted that the field has struggled to measure oligomers and hinted that a better assay might be in the works. “In the next trial we will have a more sensitive assay,” he predicted.
ALZ-801
Also at CTAD, Jeff Cummings from the Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, stressed that oligomers likely play an important role in AD pathogenesis, and may well be present throughout the course of the disease, not only in its early stages. Cummings agreed that a validated assay for Aβ oligomers in the CSF would aid development of treatments. Case in point, Alzhemed, aka homotaurine, aka tramiprosate, aka Vivimind, was recently revived once more in a new pro-drug formulation called ALZ-801. Researchers at Alzheon Inc., Framingham, Massachusetts, claim an “enveloping mechanism” by which this compound keeps Aβ from oligomerizing (Kocis et al., 2017).
In his talk, Cummings ascribed efficacy signals reported in post hoc subgroup analysis of an otherwise failed North American Phase 3 trial of tramiprosate (Aug 2007 news) to an anti-oligomer effect. Among ApoE4 homozygotes who completed the trial, the 18 patients who received the highest dose declined more slowly than those on placebo, with differences seen on the CDR-SB beginning at week 26, on the ADAS-Cog beginning at week 52, and both continuing to diverge from placebo scores through the 78-week trial.
KHK6640
While small molecules might prevent Aβ oligomers from forming or interacting with other proteins, immunotherapy is being tried in an effort to rid the brain of these nefarious peptides. At CTAD, researchers debuted an antibody developed by Kyowa Hakko Kirin, Tokyo. Perhaps better known for making Japanese Kirin beer, Kyowa is a global pharmaceutical company and has therapeutic antibodies in the pipeline for several disorders, including asthma and psoriasis. KHK6640, a humanized form of a mouse antibody, purportedly recognizes Aβ aggregates, though a search of KHK6640 on PubMed pulls up no publications.
Marc Cantillon, Kyowa Kirin Pharmaceutical Development, Princeton, New Jersey, told Alzforum that the company designed the antibody, a modified IgG4, to have little effector function to reduce the risk of ARIA. Cantillon said it was crafted to only bind an epitope on aggregated Aβ and that, unlike crenezumab or solanezumab, KHK6640 did not bind Aβ monomers.
Cantillon and colleagues detailed safety and tolerability data from a Phase 1 trial. A total of 51 AD patients with MMSEs ranging from 19.45 to 27.6 were randomized to placebo or immunotherapy in single-ascending, followed by multiple-ascending, doses. The primary outcome was safety and tolerability; pharmacokinetics, immunogenicity, and CSF level of KHK6640 were measured as secondary outcomes. Changes in CSF Aβ oligomers and cognitive scores were determined as exploratory objectives.
For the SAD study, 46 patients were randomized to either placebo or 1, 3, 10, or 20 mg/Kg of KHK6640 given intravenously, or 0.3 mg/Kg given subcutaneously. After three to 12 months, 40 of these volunteers were re-randomized to placebo or the same dose given five times at 28-day intervals. Another 11 patients who had not been tested in the SAD part were tested in the MAD study.
Overall, KHK6640 seemed well tolerated, the scientists reported. The incidence of treatment-emergent adverse events was slightly lower in the treatment groups in both SAD and MAD studies, and none of them were dose-related. Two people discontinued, one in the SAD placebo group and one in the MAD treatment group, but neither discontinuance was deemed related to drug. One person in the MAD placebo group died, and another in the MAD treatment group developed severe hypotension and was hospitalized. The hypotension resolved during the study. Nobody dropped out due to drug-related treatment-emergent events. No evidence of ARIA-E emerged, but four patients showed signs of ARIA-H, one being in the placebo group. In all cases ARIA was asymptomatic.
Pharmacokinetics showed that KHK6640 reached maximum levels in the blood within minutes, and its concentration rose with dose. Its half-life in the blood was about 17 days. Three volunteers in the SAD part and three in the MAD part tested positive for antibodies to KHK6640. Strong immunogenicity posed a problem for Eli Lilly’s pGlu-Aβ antibody LY3002813, where almost everyone who received the therapy mounted an immune response against it (Aug 2016 conference news).
In the KHK664 trial, MMSE scores trended toward higher in patients than controls, but the differences were not statistically significant and scores for the CDR and a computerized cognitive test battery were inconsistent. CSF analysis indicated that the amount of KHK6640-bound Aβ oligomers increased in a dose-dependent manner, up to 10-20 mg/kg. According to Cantillon, this may be a first AD trial to show human oligomer target engagement. The poster gave no details on how oligomers were measured.
Cantillon told Alzforum that Kirin Kyowa plans a Phase 2 global trial in prodromal AD. Details are not final yet, but Cantillon expects that outcome measures will include tau PET and scores on a cognitive composite.
Hiroyuki Shimada and colleagues from Kyowa Kirin detailed data from a similar Phase 1 study conducted in Japan. The same single-ascending doses were administered to 16 volunteers with mild to moderate AD. Again, the drug seemed well tolerated, with two drug-related treatment-emergent adverse events. Investigators concluded that one SAE, a lacunar infarction, was unrelated to the drug. No ARIA turned up in this trial, and none of the treated patients developed antibodies to KHK6640.
UB-311
Researchers at other companies are developing active vaccines to target oligomers. On a poster, Ajay Verma of United Neuroscience, Inc., Hauppauge, New York, reported data from a completed Phase 1 and an ongoing Phase 2a trial of UB-311, an active vaccine against Aβ1-14. The scientists claim this vaccine generates antibodies that bind oligomers and fibrils without causing ARIA.
Verma and colleagues used dot blots to determine antigen specificity. They reported that antibodies in serum from vaccinated patients failed to recognize α-helical monomers and Aβ monomers, but did bind Aβ oligomers and fibrils. The antibodies also bound amyloid plaques in tissue sections taken postmortem from AD brains.
While it remains to be seen exactly what these antibodies recognize, the vaccine elicited no ARIA so far in these trials. In the Phase 1, 19 people with mild to moderate AD, aged 51–78, were vaccinated; 24 weeks later, none had MRI abnormalities. Likewise, no evidence for ARIA has emerged so far in the 18–month Phase 2a study, which enrolled 43 mild to moderate AD patients, aged 60–86. They are being monitored quarterly by MRI. Twenty-five patients have completed the 12-month assessment, and none has shown signs of ARIA-E or meningo-encephalopathy. The latter sank AN1792, the first clinical Aβ immunotherapy, which was a vaccine made of Aβ1-42 (Jan 2002 news).
Crenezumab
Further along than these new immunotherapies, crenezumab is wending its way through a Phase 3 program in autosomal-dominant and early stage, late-onset AD. This antibody is said to bind oligomers, though scientists have questioned how. At CTAD, Genentech’s Jasvinder Atwal explained crenezumab’s penchant for oligomers.
Atwal reported that crenezumab binds to stable oligomers that survive passage through SDS polyacrylamide gels, whereas commercially available Aβ antibodies do not. Crenezumab also immune-precipitates high-molecular-weight Aβ species from the CSF of AD patients, and from brain extracts of PS2APP mice. In these animals, Atwal found that crenezumab, which the scientists covalently labeled with the fluorescent dye methoxy-XO4, bound to regions of the brain that are known to accumulate Aβ multimers. These include the halo surrounding Aβ plaques, and mossy fiber axons in the hippocampus. The antibody never bound to tissue from wild-type mice, which produce monomers of murine Aβ.
Perhaps most importantly, Atwal reported that crenezumab does not bind to blood vessel Aβ in the transgenic mice. “We now think that the reason crenezumab causes no ARIA is not just because it has reduced microglial effector function, but also because it fails to bind vascular amyloid,” Atwal said.—Tom Fagan
In the Running: Trial Results from CTAD Conference
Researchers presented a fall crop of Phase 1 results at the recent Clinical Trials in Alzheimer’s Disease meeting, held November 1–4 in Boston. New treatments aimed at neurotrophin signaling, tau, and cholinergic pathways moved on to Phase 2, where their sponsors face decisions about which biomarkers are worth their price tag.
LM11A-31
Sandwiched between toxic Aβ and tau proteins on one side, and synaptic dysfunction and neurite loss on the other, lies a complex web of neuronal middlemen. Think G proteins, kinases, proteases, transcriptional activators, and cytoskeletal proteins. How best to intervene when the target is not one pathway or mediator, but many? Frank Longo of Stanford University in Palo Alto, California, proposes that one answer could be LM11A-31, an activator of the p75 neurotrophin receptor. A top-level regulator of synapse life and death in the nervous system, p75’s own signaling network overlaps with those of Aβ and tau.
LM11A-31, a brain-penetrant small molecule that can be taken by mouth, boosts the pro-synaptic effects of p75 and antagonizes Aβ and tau signaling at multiple points. In animal models, LM11A counteracts the toxicity of Aβ and tau to prevent synaptic dysfunction, spine loss, neurite degeneration, microglia activation, and cognitive deficits (Knowles et al., 2013; Nguyen et al., 2014; James et al., 2017). The compound appears to reverse degeneration in 12-month-old AD mice (Simmons et al., 2014). New work presented at CTAD suggested that LM11A-31 repairs age-related loss of cholinergic neurons in wild-type mice, Longo told the audience.
Tweak a Tangled Web. LM11A-31 activation of the p75 neurotrophin receptor protects synapses by way of interfering with Aβ- and tau-induced signaling. [Courtesy of Frank Longo.]
In a Phase 1 trial, single or multiple doses of LM11A-31 taken by young or old volunteers produced no adverse events and were well tolerated. This study has now cleared the way for an international multicenter Phase 2a trial sponsored by Longo’s company, PharmatropiX, Inc., Menlo Park, California, and supported by the NIA and philanthropic funding. The trial aims to treat at least 180 mild to moderate AD patients with a low or a high dose of LM11A-31, or placebo, for 26 weeks. The primary endpoint will be safety; exploratory endpoints include a cognitive battery, CSF biomarkers, and FDG-PET.
After a low-key early development phase, LM11A-31 recently nabbed John Harrison of Metis Cognition Ltd., U.K., to help with cognitive assessments, University of Gothenberg’s Kaj Blennow for biomarker measurement, as well as Manfred Windisch at NeuroScios, St. Radegund/Graz, Austria, and Niels Andreasen and Agneta Nordberg of the Karolinska Institute in Solna, Sweden, to oversee the Phase 2a trial, Longo said at CTAD.
BIIB092
An approach to target tauopathy is advancing in the clinic, as well. Irfan Qureshi, now at Biohaven Pharmaceuticals of New Haven, Connecticut, at CTAD presented a poster on the humanized monoclonal tau antibody BIIB092. While at Bristol-Myers Squibb, Qureshi worked on this antibody, formerly known as BMS-986168. It recognizes N-terminal fragments of tau (aka eTau), which are secreted from neurons and end up in brain interstitial fluid and CSF. In mouse models of AD, the antibody lowered CSF eTau, and decreased tau-induced neuronal hyperexcitability and amyloid pathology (Bright et al., 2015). It also reportedly reduced tau toxicity in mouse models of frontotemporal dementia (Nov 2012 news). While the precise role of eTau is unclear, one idea is that it might be involved in propagating pathology from cell to cell.
Qureshi examined what ascending doses of BIIB092 would do to CSF eTau in PSP. Forty-eight participants received doses of up to 2,100 mg, infused once every four weeks for 12 weeks. The investigators saw no more adverse events in any dose than in placebo. Most events were mild, and none led to discontinuation. The antibody showed a dose-dependent accumulation in serum and CSF. Importantly, it generated a marked reduction in CSF free eTau that exceeded 90 percent for all doses. Reductions averaged 90–96 percent after 39 days of treatment, and 91–97 percent after 85 days. The investigators concluded that the antibody was safe, well tolerated, and engaged its target.
Biogen has licensed BIIB092 from Bristol-Myers Squibb, and is conducting an efficacy study in patients with PSP. The company is now developing plans for an additional Phase 2 efficacy study in Alzheimer’s disease.
BPN14770 Mark Gurney, Tetra Discovery Partners, Grand Rapids, Michigan, presented preclinical and human Phase 1 results for BPN14770, an allosteric inhibitor of the enzyme phosphodiesterase 4D (PDE4D). In the brain, PDE4D inhibitors boost cAMP, a second messenger for acetylcholine. BPN14770 hits the same pathway as acetyl cholinesterase inhibitors such as donepezil, though at a point farther downstream. Gurney and colleagues designed BPN14770 based on the PDE4D crystal structure, to try to remove a dose-limiting side effect of previous inhibitors that induced vomiting (Burgin et al., 2010).
Getting preclinical data on BPN14770 required a special effort in mouse models, Gurney said. Mouse and human PDE4D differ at a critical amino acid in the active site, and inhibitors tailored to the human enzyme are much less potent in mice. To clear that hurdle, Gurney created a mouse whose PDE4D DNA sequence was altered to match the human enzyme. Gurney showed that in these humanized mice, but not their wild-type counterparts, low doses of BPN14770 increased brain cAMP, enhanced hippocampal long-term potentiation, and improved performance in the 24-hour novel object recognition test for long-term memory. Dosing for two weeks boosted hippocampal phospho-CREB and BDNF, markers of long-term memory consolidation.
A PET ligand to PDE4D is available. In primates, it binds to brain regions important for memory and cognition, including the hippocampus and the entorhinal and prefrontal cortices. Gurney showed that BPN14770 displaced binding of this tracer, giving an estimated target occupancy rate of 83 percent for the drug in pertinent brain regions.
In a Phase 1 trial, 32 healthy volunteers received ascending doses of up to 100 mg BPN14770. Although some vomited on the 100 mg dose, there was no such distress on lower doses that still maintained pharmacologically active blood levels. Multiday dosing in both young and old volunteers produced the same pharmacokinetics, and older participants tolerated the drug well, without nausea, vomiting, or CNS effects at doses of 10, 20, or 40 mg, Gurney showed.
Even in this small trial, the drug did cause a measurable memory enhancement, Gurney claimed. Older adults treated for seven days improved their response times in the One Card Back test compared to the placebo group. The effect showed up immediately after the first treatment, and continued for the week.
BPN14770 will now advance to a Phase 2 trial in 180 patients with early AD. Gurney said he expects to see improvements in short-term, and perhaps also long-term memory. “The compound modulates a fundamental mechanism of memory. It is not amyloid-directed, and should work regardless of cholinergic deficits,” he told Alzforum.
VU319
Another cognitive enhancer entered Phase 1 testing shortly before CTAD, reported Paul Newhouse of Vanderbilt University in Nashville, Tennessee. VU319 is an allosteric modulator of the M1 muscarinic acetylcholine receptor that is being developed at Vanderbilt, with Phase 1 funding from the Alzheimer’s Association and the Alzheimer’s Drug Discovery Foundation. Newhouse showed quantitative EEG and event-related potential readouts that he will implement in the trial to judge target engagement and drug effects. He also investigates nicotinic acetylcholine activation in cognition, and recently started the Memory Improvement Through Nicotine Dosing (MIND), a two-year trial that asks whether long-term treatment with a nicotine patch improves memory and function in adults with mild cognitive impairment.
To Scan or Not to Scan …
As sponsors advance their investigational drugs, they have to decide which biomarkers to use. It can be a tough call for a small company on a budget.
Biomarkers have become de rigueur in the field; indeed, one argument holds that every drug trial should include AD biomarkers not only to better show what the drug did but also to help build a collective knowledge base tying biomarker change to disease progression and to drug response. Most researchers agree that future trial populations should be characterized with biomarkers so that results can be more fully understood. Good biomarker data can make the difference between a negative but valuable and an uninterpretable, i.e. failed, trial.
For example, adding amyloid positivity at screening to ascertain the diagnosis has become a nearly standard inclusion criterion for AD trials over the past five years. This practice followed revelations that about a third of ApoE4 noncarriers in the first solanezumab Phase 3 trials, and also in ADNI, were amyloid-negative. These participants were then thought to have had a disease other than AD.
But some scientists are pushing back. They question whether biomarkers, at least expensive ones such as PET, are always worth the expense. This is controversial among trialists, but some say the clinical diagnosis itself has improved somewhat in recent years. Take Merck’s EPOCH trial of verubecestat in mild to moderate AD (see Part 6 of this series). Its inclusion criteria did not require amyloid positivity, but even so, 90 percent of participants were amyloid-positive in both a flutemetamol PET and a CSF sub-study, each containing 21 people whose demographics were representative of the overall trial population. “This suggests that the clinical diagnosis was not so bad in this trial,” Mike Egan of Merck in Pennsylvania told the CTAD audience.
Similarly, Lon Schneider of University of Southern California, Los Angeles, reported that in a negative Phase 2 trial of edonerpic, aka T-817, 98 percent of clinically diagnosed participants turned out to be biomarker positive (see Part 12 of this series).
And Tobias Hartmann of Saarland University, Homburg, Germany, presented a new analysis done with PieterJelle Visser of Maastricht University in the Netherlands and the University of Gothenberg's Blennow, which hinted at much the same. These scientists asked how the prodromal AD population enrolled into the negative treatment trial of the medical food Souvenaid (Nov 2017 news) would have fared had it been classified with newer diagnostic criteria published after this trial started. “In this trial, most of our patients were included not based on CSF or pathology markers, but based on structural MRI. MRI was available, and participants readily agreed to do it,” Hartmann told the CTAD audience. However, many patients in this trial donated CSF, as well, and when reclassified according to new criteria that require Aβ/tau biomarker information, 91 percent of the enrolled patients ended up with the same diagnosis. “We did not expect this large overlap. I thought CSF would make more difference. That is encouraging. It is not as if you always have to have amyloid CSF or PET,” Hartmann said.
Other scientists agreed. “We can do some future mild to moderate AD trials without this expense,” said Bruno Vellas of Gerontopole in Toulouse, France. And Lawrence Honig of Columbia University in New York told Alzforum, “Maybe the clinical diagnosis is less defective than one might think. Perhaps it is not always critical to ascertain amyloid positivity, especially when it is unavailable or too costly.” —Pat McCaffrey and Gabrielle Strobel
At Least We Know These Don’t Work: Negative Trials at CTAD
Confirming once again that not only anti-amyloid therapeutic approaches struggle to score a win in Alzheimer’s and related dementias, the 10th CTAD conference, held November 1–4 in Boston, featured rest-in-peace presentations on a range of investigational drugs. They stumbled over safety in Phase 1, or fell short on efficacy in Phases 2 and 3.
Abeotaxane Adam Boxer, University of California, San Francisco, presented his center’s Phase 1 trial of TPI 287. Also known as abeotaxane, this small-molecule taxol derivative stabilizes microtubules. TPI 287 accumulates in the brain, and has been tested primarily to treat central nervous system tumors. Its application to tauopathies grew out of work showing beneficial effects of the microtubule stabilizer epothilone D in tau transgenic mice (Zhang et al., 2012). Testing of epothilone D in AD patients started in 2012 but was discontinued for lack of efficacy.
Boxer’s group examined the safety and tolerability of TPI 287 in 44 people with the primary four-repeat tauopathies cortical basal degeneration (CBD) or progressive supranuclear palsy (PSP), and in 33 people with AD. Participants received abeotaxane by intravenous infusion once every three weeks for nine weeks, with an option for open-label extension up to three months.
In recruiting for the CBD cohort, Boxer screened with amyloid PET to exclude people with AD and to limit the treatment group to people with pure tau pathology. Of 55 diagnosed with CBD, Boxer excluded seven based on positive amyloid scans. He also used CSF biomarkers to confirm diagnoses: AD patients had lower Aβ42 and higher total tau and phospho-tau levels than CBD/PSP group members, who showed elevations in neurofilament and a higher neurofilament light (NfL)/phospho-tau ratio than the AD group.
Participants received tailored doses of 2, 6.3, or 20 mg/meter2 TPI 287, or placebo.
AD patients tolerated the treatment poorly. Boxer told the CTAD audience that he had to stop the high-dose arm because two participants suffered anaphylactoid hypersensitivity reactions, most likely to the diluent for the active compound. In all, seven people in the AD group discontinued treatment. Curiously, the CBD/PSP group tolerated the drug well, even at the highest dose. They suffered no hypersensitivity reactions, and most participants stuck with the trial even through the open-label extension. However, in CBD and PSP patients, the drug caused more falls, a serious concern.
On the exploratory cognitive endpoints, the researchers saw a hint of stabilization of MMSE scores in the AD group, but no change in the ADAS-Cog, and the CBD/PSP cohort had a dose-related worsening on the Clinical Dementia Rating-sum of boxes at three months.
Boxer has no future plans for the drug, except to complete the analyses of pharmacokinetics and MRIs. He told Alzforum that investigators learned a lot from the trial. “It shows the importance of testing potential treatments in different tauopathies," he said. “Animal models don't tell the whole story, and we have to look at different conditions in humans," he said.
Edonerpic
Previously known as T-817, this compound is thought to protect neurons, possibly by activating sigma receptors. An earlier Phase 2 trial had a difficult time recruiting and high dropouts, yet the trial’s overall null result looked to the sponsor, Toyama Chemical, as if a treatment benefit for more advanced patients might be had with higher doses. The company contracted the Alzheimer’s Disease Cooperative Study (ADCS) to run another trial. At CTAD, Lon Schneider of the University of Southern California, Los Angeles, presented results of that trial.
In short, this Phase 2 study also delivered a null result, where the treatment failed to outperform placebo on the primary outcome, the ADAS-cog, or any of the secondary outcomes. On the upside, Schneider told the audience, “We can now trust this result because this was a well-designed and well-run study.”
For one, the trial included frequent assessments to make the outcome measurements more precise. For another, even though the trial did not require biomarker positivity as an inclusion criterion to ensure participants truly had AD, CSF samples taken for exploratory measurement of edonerpic’s effects on Aβ and tau confirmed that 98 percent of the study participants indeed were positive for the CSF AD signature at baseline. In other words, the clinical AD diagnosis used in this trial worked.
Conducted at 52 sites, the trial enrolled 476 people with a diagnosis of probable AD and an MMSE of between 12 and 22, and randomized them to either placebo or 224 mg or 448 mg of edonerpic once daily for a year. Outcomes were measured at 12, 24, 36, 44, and 52 weeks. MRI of the whole brain, ventricles, and hippocampus, and CSF measurement of Aβ40/42 and total and phospho-tau formed part of the trial.
Dropouts in the placebo, low dose, and high dose were 11.4, 29.5, and 24.1 percent, respectively. Some biomarkers showed changes but the changes did not all trend in the same direction. For example, CSF p-tau dropped on the high dose, hippocampal shrinkage slowed on the low dose, and different MRI analysis techniques yielded different results. “We need better neurodegeneration markers when we evaluate drugs that are presumably neuroprotective,” Schneider said.
S47445
This drug is a positive allosteric modulator of AMPA glutamate receptors. The French company Servier has been evaluating it for people with Alzheimer’s disease who also have symptoms of depression (see Therapeutics database). At CTAD, Maria Pueyo told the audience that Servier decided to end its work with this drug in Alzheimer’s. That’s because its latest, six-month, Phase 2 trial of 520 people with mild to moderate Alzheimer’s showed no significant differences between the drug and placebo groups on the primary outcome, the ADAS-cog, or on secondary measures of daily function or depression for either of the three doses used.
“Safety was good but we had no efficacy on cognition or function,” Pueyo said. The drug did get into the CSF and increased glutamate in the brain, Pueyo said in response to audience questions, but still achieved none of the desired benefit. Curiously, symptoms of depression improved in both drug and placebo groups, perhaps as a consequence of receiving the added care and attention that come with participating in a clinical trial.
Tricaprilin Samuel Henderson of Accera Inc. in Boulder, Colorado, explained negative results for tricaprilin, Accera’s latest formulation of caprylic triglycerides to boost ketone body metabolism with a dietary aid. Nourish-AD was a Phase 3 trial conducted at 61 sites in the U.S., in 413 people who met the NINDS-ADRDA diagnosis of probable AD.
At the end of the trial, the treatment and placebo groups performed equally on the primary outcome measure, cognition as per the ADAS-cog, and the secondary outcome measure, overall function as per the ADCS-CGIC. As with prior formulations of this dietary approach, adverse events included nausea, vomiting, and gastrointestinal discomfort.
At CTAD Henderson ascribed the negative result to low exposure and bioavailability, saying this new formulation generated insufficient amounts of ketone bodies in the brain. As happens frequently in six-month trials in AD, the tricaprilin trial, too, was further hobbled by a relative lack of cognitive decline in the placebo group.
Henderson noted the company still has faith in the rationale behind ketone body therapy for treating neurologic disease, and has developed yet another formulation for its approach.—Pat McCaffrey and Gabrielle Strobel
No Negativity, Please. Clinical Trial Leader Urges Focus on Learning, Progress
In 2016, Rachelle Doody switched from academia to industry when she left Baylor College of Medicine for Roche/Genentech in Basel, Switzerland. At this year’s Clinical Trials on Alzheimer’s Conference, held November 1–4 in Boston, Doody used a keynote address to call for more balanced reporting of clinical trials in Alzheimer’s disease, and for more optimism all around.
After four cholinesterase inhibitor drugs were approved between 1993 and 2001, AD trials research entered a dry spell that was tallied and highlighted recently in a review article. In it, the authors counted 413 trials of 244 unique compounds tested between 2002 and 2012, of which only Namenda—an old drug previously in use in Europe—garnered FDA approval (Cummings et al., 2014). This amounted to an overall failure rate of 99.6 percent, which was widely reported in the press and indeed keeps haunting the field of AD research. “The media really keyed in on that 99.6 percent number,” Doody told the CTAD audience. She quoted Fox News, BBC News, and Scientific American; Fortune, Genetic & Engineering News, FierceBiotech, and other outlets had also reported it.
Painting AD research with a broad, negative brush is at odds with a general sense of scientific progress among the research community; what’s worse, a “failure number” creates a downward spiral, Doody cautioned. In media stories citing this number, the general public hears that Alzheimer’s is hopeless and trial participation pointless. Academic investigators see a devaluation of their work on shared multicenter trials. Within industry, overly negative headlines drive down internal calculations of a metric called probability of technical success. The PTS, in turn, influences another internal metric called net present value, by which a given investigational AD therapy competes for investment with candidate drugs in other disease areas perceived to be “easier” or more likely to generate marketing approval, such as cancer.
Citing 2016 numbers from the Biotechnology Innovation Organization (BIO), Doody acknowledged that far fewer investigational drugs progress through the clinical trial phases and to regulatory approval in Alzheimer’s than in the pharmaceutical industry overall. The reasons, however, are manifold and changing.
For example, comparing oncology, a field that has seen much progress in the past decade, with AD, Doody noted that AD trials are not only fewer but also far larger. An AD trial enrolls hundreds to thousands of participants in Phases 2 and 3, respectively, versus an average 20 to 30 participants per oncology trial in all phases. Moreover, AD drugs span a range of targets and approaches, from active and passive immunotherapies, various receptor agonists or antagonists, inhibitors of different enzymes, to growth factors, repurposed drugs, hormones, even nutraceuticals. Each one of these had its own reason for having failed, and distilling them all into a single failure number is counterproductive, Doody argued.
Both internal postmortems and reporting to the general public should focus on what each failure has taught the field, and what is being done differently in the next attempt. Some drugs failed because they never reached their pharmacodynamics targets, or the trial included no robust measure of target engagement in the brain. In other trials, the mechanism of action was vague or not central to AD progression. Some drugs foundered under dose-limiting side effects, others because their studies were statistically underpowered. Some trials stumbled because the placebo group did not decline or large variability between the participating sites precluded analysis. Yet other drugs were tested in heterogeneous, insufficiently characterized patient populations. And so on. The point is that the field is learning from every trial setback, Doody said.
So what has been learned? For one, it appears that drugs that modify risk factors of AD belong in prevention trials. Drugs targeting neuropathology, which begins years before symptoms start, belong in very early-stage trials. For another, it appears that the most established Aβ/tau CSF biomarkers help identify patients with AD pathology but poorly predict progression or measure drug response. The field needs additional markers that change dynamically as people develop symptoms and get worse; for this purpose, research-grade tests, such as neurofilament light, neurogranin, and tau PET, are coming along rapidly.
And what is different today? Many aspects of AD trials are changing. New trials incorporate criteria for prodromal AD, anti-amyloid trials verify amyloid positivity for inclusion and increasingly incorporate PET as outcome measures, and tau PET is entering therapeutic trials. Trialists and regulators together agree that, for the time being, trials at the prodromal stage won’t aim for surrogate biomarkers as approvable outcome measures, but instead will work out sensitive measures of subtle cognitive decline (see Part 1 of this series). Regulators support the field’s deepening understanding of the AD preclinical period by focusing on composite measures and accepting a single Phase 3 trial.
On anti-amyloid antibodies and BACE inhibitors, at least it is now clear that they can remove amyloid from the human brain, or slow its accumulation there. While this has not helped people with symptomatic AD, earlier intervention may well do so, Doody said.
She called on her colleagues in both academia and industry to include lessons learned when discussing a negative trial, and to encourage both investment and enthusiasm for new trials. “We need to work together to send a positive message to patients about participating in clinical trials. We also need to help trial sites maintain respect for what they are doing, and to offer clinical trials as options for all patients,” Doody concluded. —Gabrielle Strobel


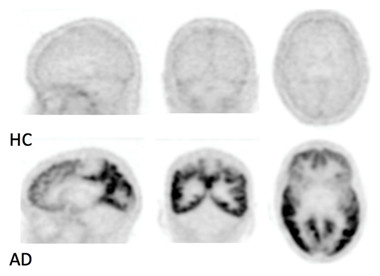



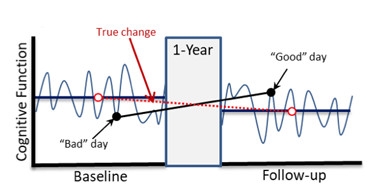












Comments
No Available Comments
Make a Comment
To make a comment you must login or register.