Clinical trials, brain imaging, biomarkers, and intervention studies drew more than 4,000 researchers to the Alzheimer's Association International Conference 2014 at the Copenhagen Congress Center in Demark. Data from Phase 2 clinical trials of crenezumab, an anti-Aβ antibody, disappointed, but positive results from a large Finish intervention study raised hope that lifestyle changes can pay dividends toward preventing dementia. The Alzheimer's Prevention Initiative announced a new partnership with Novartis to test if an active Aβ vaccine or a BACE inhibitor can slow or prevent sporadic Alzheimer's in people who are genetically predisposed to the disease. Abstracts for the meeting are freely available here.
Novartis to Partner with Banner Health on ApoE4 Prevention Trial
At the Alzheimer Association International Conference in Copenhagen, Denmark, on July 15, researchers from the Banner Alzheimer's Institute in Phoenix introduced Novartis Pharmaceuticals Corporation as their partner in the upcoming Alzheimer's Prevention Initiative ApoE4 trial. Banner will work with the Swiss pharmaceutical company to test experimental therapies in people who carry two copies of the ApoE4 allele, the major genetic risk factor for late-onset Alzheimer's disease. The study complements the API ADAD trial, which will test the anti-Aβ antibody crenezumab in otherwise healthy people who carry an autosomal-dominant AD mutation and typically begin to show signs of the disease by age 50 (see May 2012 conference news). "We are excited about the chance to evaluate promising preclinical AD treatments in those at risk for developing AD at older ages," said Eric Reiman, who leads API together with Pierre Tariot and Jessica Langbaum at Banner. "We previously argued that now is the time to launch a new era in Alzheimer’s prevention research—one in which we could efficiently test the range of preclinical AD treatments and find those that work,” he said. Reiman told Alzforum that he believes that new era has arrived.
Since the API APOE4 trial was announced, researchers in the field have wondered who the industry partner would be and what intervention would be tested (see Sep 2013 community news). The choice of Novartis came after a long vetting process. It provides the opportunity to test two anti-amyloid treatments, rather than the one originally envisaged, in a study that is about twice as large as originally planned. Cognitively normal volunteers aged 60 to 75 will be randomized to placebo, an Aβ vaccine, or a β-secretase inhibitor. Full details are yet to emerge. CAD106, Novartis' active Aβ immunotherapy, has completed Phase 2 testing. The company's vice president, Michael Ryan, told Alzforum that Novartis has a BACE inhibitor poised to enter Phase 1 testing.
API APOE4 plans to recruit more than 1,300 volunteers in North America and Europe. Only 2 percent of the world's population carries two copies of the ApoE4 allele. The trial will recruit partly through API's Prevention Registry, which has 39,000 members and plans to enroll up to 250,000. Banner estimates the active immunotherapy part of the trial will begin in late 2015 and the BACE intervention a year later. Each part of the trial will run for five years. Each of the two treatment arms are expected to enroll 390 volunteers and each placebo arm 260, but the exact number could change based on regulatory agency input.
Reiman and colleagues are considering change in cognition judged by the API composite cognitive score as the primary outcome measure, although time to clinical onset is a possibility as well. The trial will analyze a suite of biomarkers, including brain Aβ and tau, through imaging and cerebrospinal fluid analysis, glucose metabolism via FDG PET, and structural imaging via MRI. There will be interim testing at two years as well as at the beginning and end of the study. Evaluating how biomarkers change in relation to clinical benefit (theragnostics) will be an important part of the trial, said Reiman, since that information will be crucial to validating biomarkers as endpoints in future prevention trials.
Cognition is likely to be tested more frequently. Reiman and Tariot stressed that full protocol is still under discussion and has yet to be submitted for regulatory approval. API APOE4 will also assess the short- and long-term impact of disclosing ApoE4 status. Potential volunteers must consent to genotyping and can avail themselves of genetic counseling provided by the trial. The infrastructure established as part of this trial will also serve as a platform for future API trials in persons at risk for late-onset AD, said Reiman.
The API ADAD trial was estimated to cost more than $100 million. The price tag for the much larger API APOE4 trial was not disclosed, but $33.2 million comes from the National Institutes of Health, $15 million from the Banner Alzheimer's Foundation, and additional funding will come from Novartis. Other private foundations and the Accelerating Medicines Partnership, which supports research into biomarkers and therapies for AD, will make up the balance (see Feb 2014 community news).
In keeping with the API philosophy of openness, Novartis has agreed that all data and samples will be shared with the research community once the trial finishes.—Tom Fagan
Crenezumab Disappoints in Phase 2, Researchers Remain Hopeful
The anti-Aβ antibody crenezumab missed both its primary endpoints in two Phase 2 clinical trials, according to results presented at the Alzheimer’s Association International Conference 2014, held in Copenhagen July 12-17. The ABBY trial fell short on goals set for cognition and function. The smaller BLAZE study, which was designed to measure biomarker effects, showed no effect on cognition as a secondary endpoint. Results of both trials indicated that the drug was safe and well-tolerated. Post-hoc analysis of the data, presented by Jeffrey Cummings, Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, hinted that the drug may yet slow decline in patients with mild AD, echoing the outcome of trials of solanezumab, an anti-Aβ antibody being developed by Eli Lilly (see Oct 2012 news story).
Perhaps because researchers in the field have grown used to negative clinical trial data, the news seemed anticlimactic, sparking little debate from the packed hall at the Bella Center in Orestad, just north of the Danish capital. In interviews with Alzforum, some researchers lamented the practice of slicing and dicing data to find significant effects in ever-smaller patient groups, while others noted that this was an opportunity to learn and to design a Phase 3 trial to test if the treatment works.
Crenezumabis a monoclonal antibody developed by AC Immune and licensed to Genentech. It binds all forms of Aβ, including monomers, oligomers, and fibrils. "Of all the immunotherapies currently in trials, I thought crenezumab might have the best chance of working," Dave Morgan, University of South Florida, Tampa, told Alzforum. "It not only binds and helps remove plaques, but also blocks Aβ aggregation at substoichiometric amounts." Crenezumab was designed to limit inflammatory responses while still promoting uptake of Aβ by microglia (see Jul 2012 research news).
In ABBY, 122 patients with mild to moderate Alzheimer’s were randomized to 300 mg of crenezumab injected subcutaneously every two weeks, while 62 volunteers got placebo. Another 163 patients received 15 mg/kg of the antibody intravenously every four weeks and 84 were given a placebo. The IV method delivered about twice as much antibody as the subcutaneous route over the 68-week course of treatment. No overall difference emerged between the placebo and treatment groups on cognitive co-primary outcomes measured by the ADAS-Cog12 and the CDR sum of boxes. Daily function, as determined by the ADCS Activities of Daily Living, declined as quickly in patients taking the drug as in those on placebo. In prespecified analysis of patients with an MMSE score of 20-26, i.e. the milder subgroup, the higher IV dose tended to slow cognitive decline, but the results missed statistical significance. A second, exploratory analysis of the 121 patients with even milder AD, an MMSE of 22-26, showed a trend toward efficacy at 49 weeks and a statistically significant effect at the end of the trial.
The smaller BLAZE trial enrolled 91 patients with mild to moderate AD who tested positive for brain amyloid on a Florbetapir PET scan. Here too, low and high doses of crenezumab were given via subcutaneous (25 treatment, 13 placebo) and intravenous routes (29 treatment, 15 placebo). The result on PET amyloid imaging—the primary outcome of BLAZE—will be presented at the Clinical Trials on Alzheimer's Disease conference in November, said Cummings. The higher dose of crenezumab trended toward preserving cognition in this trial.
"We believe that this was a very good outcome for a Phase 2 trial designed and powered as it was," Andrea Pfeifer, CEO of AC Immune, told Alzforum. "We believe showing a signal and target engagement is a positive outcome for a trial that was not designed to be a pivotal one," she said. Pfeifer stressed that it was important to estimate a treatment effect in Phase 2, because this provides a baseline from which to power a Phase 3 trial. Rachelle Doody, Baylor College of Medicine, Houston, told Alzforum she was concerned that many people, particularly in the media, were jumping to the conclusion that crenezumab was doomed, looking at the data as if it were a Phase 3 trial. "They are trying to emphasize a message that is at best a small piece of what was learned, and at worst a distortion," she said.
What was learned? "About all we can say for sure about these findings is the drug appears safe," said Todd Golde, University of Florida, Gainesville. Similar sentiments came from Eric Reiman, Banner Alzheimer's Institute, Phoenix. "The findings show [crenezumab] failed to meet its primary endpoints; beyond that, interpretation would be preliminary," he said. Doody called the parallels between the crenezumab and solanezumab data thought-provoking. In both, ADAS-Cog trends showed separation between placebo and treatment groups that grew over time. Both treatments target soluble Aβ. "While we have to wait for the biomarker data, if both [solanezumab and crenezumab] have similar signals, it makes you wonder if it is necessary to target fibrillar Aβ," she suggested (see Apr 2013 conference news). Doody has consulted for many pharmaceutical companies, including Roche, Genentech, and AC Immune, but not for Eli Lilly.
Reiman, who co-leads the Alzheimer's Prevention Initiative, also commented on the similarities to the solanezumab data. The results have bearing on the API’s ongoing trial of crenezumab in families carrying an autosomal-dominant presenilin mutation (see Mar 2011 research news). "We are encouraged by the continued safety and tolerability, which is very important in healthy volunteers, and that the pattern looked strikingly similar to what Lilly reported for solanezumab," Reiman told Alzforum. "API is predicated on the idea that introducing treatment early will have a more profound effect. If that turned out to be confirmed, it would be very encouraging."
The API trial uses the subcutaneous regimen, which showed no cognitive benefit in ABBY, even in the post-hoc analysis. “Based on these findings we will discuss with health authorities the possibility of increasing the subcutaneous dose leading to comparable concentrations as in the IV dose,” Reiman told Alzforum. Pfeifer noted that when treating a healthy population, it could be assumed that a lower dose may be needed than in a treatment trial. She said she had no doubt the API trial will continue.
On the separate question of whether Genentech will launch new clinical trials of crenezumab based on these results, a company spokesperson stuck to this formal statement, “We are continuing to further analyze the Phase 2 data and will be meeting with health authorities, including the FDA and the EMA, in 2014. It is premature for us to discuss the development plans for crenezumab.”—Tom Fagan
Could the eyes be a portal by which to check for Alzheimer’s pathology in the brain? Some researchers at the Alzheimer’s Association International Conference 2014, held July 12-17 in Copenhagen, believe so. Two groups presented results about detecting Aβ in the eye. One team reported that Aβ in the retina correlates with plaque buildup in the brain, while another has similar results from the lens. Both propose to develop these biomarkers as early screening tests for Alzheimer’s disease. “PET amyloid imaging and magnetic resonance are expensive,” said David Knopman, Mayo Clinic, Rochester, Minnesota, who was not involved in either study. If they pan out, these developing techniques may prove “simpler, less invasive, and more feasible to use in a primary-care setting as opposed to a large research center.”
Aβ in the Retina Shaun Frost, from the Commonwealth Scientific and Industrial Research Organization (CSIRO), Perth, Australia, views the retina as an extension of the brain that provides an opportunity to diagnose and track progression of Alzheimer’s. This layer of nervous tissue at the back of the eye has a similar vasculature to the brain and connects directly to it through the optic nerve. Frost presented preliminary results on a test that he says picks up Aβ plaques in the retina. Participants drink a proprietary formulation of curcumin, a component of the spice turmeric. The compound crosses the blood-retinal barrier, binds to amyloid plaques, and fluoresces, so researchers can detect it with a specialized camera developed by collaborator NeuroVision, a company based in Sacramento, California. Aβ plaques appear as fluorescent spots on the retinal scan (see image below). Frost’s colleagues have conducted experiments on postmortem human retinas and reported that curcumin binds to plaques, Frost said (see Koronyo-Hamaoui et al., 2011).
Plaques tagged by curcumin fluoresce in a retinal scan. [Image courtesy of NeuroVision Imaging.]
Frost noted that retinal scans provide 100 to 1,000 times finer resolution than PET scans—down to microns, rather than millimeters. This means researchers can identify individual plaques and monitor their changes.
Thus far, Frost and colleagues have imaged the retinas of 40 of the 200 patients they plan to test from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). They were on average 73 years old and had either AD, mild cognitive impairment (MCI), or were healthy. All had undergone a PiB PET scan. In this cross-sectional study, retinal and brain amyloid had a coefficient of correlation of 0.76. Everyone who tested positive in PiB PET, including healthy controls, also showed evidence of plaques in the retina, meaning there were no false negatives. “That’s a crucial component of a screening test for Alzheimer’s because you don’t want to leave any [positive individuals] behind,” said Frost.
However, false positives did occur. Some people whose PiB PET was negative nonetheless tested positive in the retinal scan. Specificity compared to brain imaging was 85 percent. If larger studies bear this out, it might mean that the new technique detects plaque formation prior to PiB PET, suggested Frost. Longitudinal data will be required to find out.
Longitudinal work on this technique is starting now. One MCI patient imaged twice in three months showed a 3 percent increase in retinal plaque, suggesting the technique may eventually help monitor small-scale changes over time, Frost said. Since this method involves no radiation, it could be performed more frequently than PiB PET. It may help determine if candidate drugs remove plaque in clinical trials on a short time scale, said Frost. This technique could also cheaply select people with evidence of AD-related pathology for clinical trials. Some researchers are expressing interest in including these measures in upcoming clinical trials, Frost claimed.
Researchers seem intrigued by the work. One asked if tau could be imaged the same way, and Frost said this has not been addressed yet. Another scientist wondered if fatty deposits called drusen, which associate with age-related macular degeneration, could be confused with Aβ deposits. Frost replied that drusen occur in the macular region in the retina’s center and in its deeper layers, whereas amyloid plaques are peripheral and shallower; therefore the two are easy to differentiate. Jochen Herms, Ludwig Maximilians University, Munich, noted the retina’s known autofluorescence. He talked of having tried time and again in vain to find evidence of Aβ in the retina in mice or in humans, and questioned how researchers can see plaques against background noise. Frost responded that he takes a baseline image before dosing with curcumin and pointed to a publication that reported evidence of retinal amyloid (see Alexandrov et al., 2011).
What about the lens?
Aβ was first reported in postmortem lenses of AD patients more than a decade ago. Lee Goldstein of Boston University, Massachusetts, reported that Aβ40 and Aβ42 accumulated in lens fiber cells (see Goldstein et at al., 2003). Since then, many researchers have tried but failed to reproduce the finding, though others are convinced the Aβ deposits are there and may provide an in vivo biomarker for AD (see May 2013 news story).
Paul Hartung, president and CEO of Cognoptix, Inc. (formerly Neuroptix), in Acton, Massachusetts, presented results from the company’s Phase 2 trial of its Fluorescent Ligand Eye Scanning (FLES) technique, which reportedly detects Aβ in the lens. In FLES, an ointment is applied to the eye the night before a scan. The formulation contains a small fluorophore that diffuses into the eye overnight and binds to Aβ. The next day, a harmless laser delivers quick pulses of light to the eye and detects bound compound by way of its fluorescence.
Hartung said that the method was safe and results correlated with florbetapir amyloid PET scans. In 20 patients with probable AD and 20 age-matched healthy controls, the researchers determined who was positive for amyloid based on a threshold of the fluorescence uptake value. Hartung claimed that with a sensitivity of 85 percent and specificity of 95 percent, lens positivity correlated better to the clinical diagnosis than did brain PiB PET. “We see the potential for this technology to aid in diagnosis and clinical staging of disease,” he said.
Researchers at the meeting asked how the Aβ plaque gets into the lens, which has no blood vessels. Hartung cited evidence that the amyloid precursor protein, secretases, and Aβ-degrading enzymes are present in the lens (see Li et al., 2003); he believes the deposits are likely manufactured in the lens. Someone asked if Cognoptix had correlated what they found with biochemical analysis of lenses removed in surgery or from cadavers. Hartung responded that they had not, but that Goldstein's original study noted the presence of deposits in postmortem lenses.
Scientists were generally skeptical about this technique. Some pointed out that the idea for a lens biomarker has been around for years, yet little has come of it. Others said that since the lens is not directly connected to the brain, it might not reflect brain processes. “The lens is independent of the brain,” Creighton Phelps, National Institute on Aging, Bethesda, Maryland, told Alzforum. “It is difficult to understand how amyloid accumulation in the lens reflects changes in the brain related to Alzheimer’s disease.” Kaj Blennow, University of Gothenburg, Sweden, who chaired the session, agreed, noting that the lens contains no blood vessels, neurons, or nerve terminals. “It is unclear why it would have the same type of pathology as in the brain,” he wrote to Alzforum in an email. “Solid biochemical data showing that there are Aβ deposits in cases with a positive [amyloid imaging] test would be highly desirable.” Others said that if the test works, then conceptual arguments become moot. “If the lens truly accumulates Aβ, then the question is simply how tightly does it mimic brain amyloid levels?” Knopman said. He suggested Hartung and colleagues test the idea in larger, diverse populations.
Many researchers at the conference noted that to their minds, it makes more immediate sense to seek amyloid in the retina, although Blennow maintained that it seems illogical to look for pathology in an organ that is unaffected by symptoms of Alzheimer’s. Frost pointed out that even if a treatment cleared plaque from the brain, it likely would not clear it from the lens, meaning it would not help monitor treatment responses in the brain. The focus of that work is more on differential diagnosis, he said. For his part, Hartung responded that pathologies brought on by macular degeneration or ocular amyloidosis might confound results in the retina.—Gwyneth Dickey Zakaib
References and Thresholds—Amyloid Imaging Protocols Debated at AAIC
Compared to tau imaging, PET scanning for amyloid may seem like ancient technology, but there is ample room for improvement. At the Alzheimer's Association International Conference 2014, held July 12-17 in Copenhagen, Denmark, researchers proposed changing imaging protocols to sharpen their diagnostic and prognostic potential and improve the power to track amyloid changes over time. The change might require re-analysis of some existing data sets.
In a poster presentation, Kewei Chen from Banner Alzheimer's Institute, Phoenix, made the case for using cerebral white matter as a reference region for Florbetapir PET imaging of brain amyloid. Most labs now use the cerebellum or pons when calculating standard uptake value ratios (SUVRs) to quantify Aβ deposits. As co-author Eric Reiman explained, much of the cerebellum and pons lie below cerebral sites where amyloid accumulates, such as the entorhinal cortex, hippocampus, and medial frontal cortex (see image below). As PET scanners "slice" through the brain, the cerebellum and pons rarely fall in the same plane as Aβ-positive tissue; in fact they lie at the bottom of the field of view of the PET camera, where a slight change in head position could introduce variability from scan to scan. "We think that scans taken lower in the brain introduce a lot of noise," said Reiman.
Location, location, location.
Cerebral white matter (yellow) makes a better reference for cerebral (gray) Aβ than the cerebellum (purple) or the pons (green), which lie below sites of mean cortical uptake of the amyloid tracker flobetapir (red). [Image courtesy of Kewei Chen, Banner Health.]
Chen and colleagues wondered if a reference region higher in the brain would improve the power to track amyloid change over time. They re-analyzed data from the Alzheimer's Disease Neuroimaging Initiative comparing the cerebellum, pons, and cerebral white matter as reference regions. Chen correlated the change in cortical SUVRs over two years to cognitive decline among 332 people (31 with probable AD, 187 with MCI, and 114 cognitively normal controls). Only SUVR changes calculated using white matter correlated with deterioration on the MMSE, ADAS-Cog, and CDR sum of boxes. Moreover, SUVRs calculated based on the reference regions varied less from person to person. For example, among people with AD who tested positive for amyloid at baseline, only the white matter reference method showed consistent increases in amyloid deposition in the cortex over the two years. Values based on the cerebellum and the pons fluctuated and suggested that some patients actually lost amyloid.
"The field is slowly coming around to the idea that for longitudinal measurements, white matter gives better signal and less noise," said William Jagust, University of California, Berkeley. In a pre-meeting imaging conference, Jagust reported highly variable changes in amyloid load among ADNI volunteers who started out with similar Aβ levels. That was partly due to problems calculating SUVRs. "The reference region was a real headache," Jagust said. His colleague Susan Landau, also from Berkeley, used the cerebellum as a reference, but that was suboptimal, said Jagust. Other regions, including the brainstem, the pons, and white matter, gave more consistent results.
Other groups are noticing similar trends. At AAIC, researchers from Avid Radiopharmaceuticals in Philadelphia reported that using white matter as a reference region increased the signal-to-noise ratio when measuring longitudinal changes in ADNI participants. In a poster, first author Abhinay Joshi showed that among amyloid-positive volunteers, baseline SUVRs correlated more tightly with two-year values when referenced against white matter (r=0.9) than cerebellum (r=0.73). The upshot was that for measuring change, white matter makes a better reference.
Does changing the reference region matter? Based on the two-year ADNI data, Chen and colleagues calculated how many patients might be needed to measure a 25 percent reduction in brain amyloid in a clinical trial. While about 8,000 Aβ-positive MCI patients would have to be enrolled to see an effect over one year using the cerebellar reference, 325 would suffice if the white matter was used instead. "The differences are staggering," said Reiman. "We believe that the use of a cerebral white matter reference region will greatly improve the possibility to track longitudinal changes in fibrillar amyloid PET measurements, relate them to clinical decline, and evaluate amyloid-modifying treatments in clinical trials." While additional studies are needed to clarify whether the findings apply to different amyloid PET ligands and PET scanners, Reiman suggested that researchers might want to re-analyze previous trial data with this approach.
Yet another measure that colors interpretation of imaging data is the threshold researchers use to classify a person as amyloid-positive or -negative. Many groups set an SUVR value of 1.4 or higher as being amyloid positive. In the preconference imaging symposium, Sylvia Villeneuve, who works with Jagust at Berkeley, claimed this number is too high. In an approach others praised for its elegance, she reanalyzed the rationale for this figure and concluded that it should be lower.
First, Villeneuve compared amyloid uptake among 154 cognitively normal old adults and a sample of young adults presumed to be amyloid-free. That analysis suggested that a Pittsburgh compound B (PiB) SUVR threshold of 1.19 predicts amyloid deposition. In another test, she looked at Gaussian distributions of SUVRs. That showed two major groups with low and high values that reflect amyloid-negative and -positive individuals. An SUVR of 1.22 equated to a 90 percent probability of being in the low or amyloid-negative range. She obtained the same value from a cluster analysis, where she looked at 74 different brain regions to see which discriminated individuals with and without amyloid and at what SUVR threshold amyloid began to appear.
Finally, Villeneuve reported a voxel-wise estimate. In this analysis, she took 22 people with the lowest SUVRs, compared them to 22 people with the next-highest SUVRs (the test group), and looked voxel by voxel for differences in amyloid. If none appeared, she added the person with the next-highest SUVR into the test group (removing the person with the lowest SUVR) and re-ran the analysis. She kept this "sliding-window approach" going until she detected a difference between the two groups. That occurred when the SUVR reached 1.19.
Villeneuve used this data to suggest that lower thresholds would better capture people who are Aβ positive. Cliff Jack, Mayo Clinic, Rochester, Minnesota, was concerned about false positives and said that lower thresholds might only work for specific amyloid ligands and specific algorithms. "If we used a threshold of 1.2, everyone over 50 would be amyloid-positive," he said.
Jagust, working with Gil Rabinovici at the University of California, San Francisco, correlated thresholds with pathological data from about 50 cases of dementia/MCI and a few controls. At AAIC, he reported that the lower threshold SUVR values for PiB are highly specific. "We were surprised it did not lead to more false positives," he said. On top of this, the sensitivity increased to 80 percent, from 60 percent for the high-threshold SUVR. "The literature suggests sensitivity is mid- to high 90s for the higher-threshold values," he said. When he applied the higher SUVRs reported by others, the sensitivity was lower. "If we use the high-threshold values, we may miss a fair proportion of cases that have low numbers of plaques," he said.
Jagust agreed with Jack that many different parameters go into setting SUVR thresholds. The important thing is to pick the number rationally, not just follow figures reported in the literature. "If you want to find out how amyloid changes as people age, for example, then you may want to use a lower threshold," he said.
Victor Villemagne, Austin Health in Melbourne, Australia, used Villeneuve's lower SUVR threshold to estimate how long it takes people to develop memory problems once they become amyloid positive. He reported that in the Australian Imaging Biomarkers & Lifestyle Flagship Study of Ageing, memory impairment correlated with SUVRs of 1.4 and above. Based on a pathology threshold of 1.2 and a memory threshold of 1.4, Villemagne calculated a window of five to seven years’ time to intervene before people with the first wisps of amyloid in the brain start to notice memory problems.—Tom Fagan
Anti-Amyloid Therapies Combine Forces to Knock Out Plaques
Puny efficacy signals of monotherapies and a growing awareness of the complexity of Alzheimer’s disease have made combination therapy a hot topic in the past few years. However, researchers are only beginning to explore this strategy in animal models. At the Alzheimer’s Association International Conference 2014, held July 12-17 in Copenhagen, Denmark, Ron DeMattos of Eli Lilly and Company, Indianapolis, Indiana, presented data on his company’s first attempt. In aged PDAPP mice, investigators found that combining a BACE inhibitor and a pyroglutamate Aβ antibody cleared plaques more thoroughly than either therapy alone over a four-month period. Statistics indicated a synergistic effect, meaning that the combination cleared more plaque than the single effects added together. “This provides a strong rationale in terms of moving toward the clinic with this approach,” said DeMattos.
Other scientists were impressed by his talk. “I thought this was a spectacular piece of science,” said Eric Karran, Alzheimer’s Research UK, Cambridge. “DeMattos revealed in an exquisitely compelling way that when you combine a BACE inhibitor with a specific antibody that will target the plaque, you can have very dramatic effects on plaque loads.”
The mE8 antibody (blue) targets deposits (round, yellow), while a BACE inhibitor reduces production of Aβ peptides (yellow rods).
[Image courtesy of Neuron, DeMattos et al., 2012.]
To date, the only other study examining a combination therapy came from scientists at F. Hoffmann-La Roche Ltd. in Basel, Switzerland. They demonstrated the additive effects of the company’s discontinued BACE inhibitor R7129 and its Phase 3 antibody gantenerumab in reducing Aβ levels and plaque from transgenic mice (see April 2013 news story).
DeMattos and colleagues took a similar approach. They shut off Aβ production with the LY2811376 BACE inhibitor (see May et al., 2011), while clearing existing Aβ aggregates with mE8, an anti-pyroglutamate Aβ antibody that is specific to plaque (see DeMattos et al., 2012).
Lilly scientists assigned 180 18-month-old PDAPP mice to one of five cohorts. The first was sacrificed to determine the levels of plaque in the brain at baseline. The other four cohorts served as the control group, mE8-treated group, BACE-inhibitor treated group, and combination group. Treatment lasted for four months, with BACE inhibitor given through food and mE8 injected subcutaneously each week. At the end of the study, the researchers extracted Aβ biochemically and used quantitative ELISA to measure the mass of the Aβ deposits in the hippocampus and cortex.
In the control group, plaque load in the hippocampus had doubled from baseline. The mE8- and BACE inhibitor-treated groups had 49 and 66 percent less plaque, respectively, than controls. In the combination group, amyloid levels dropped by 86 percent. Statistical analysis suggested the treatment effects were additive, with trends toward synergy.
Staining brain slices of the hippocampus and cortex with 3D6, an Aβ antibody that visualizes both diffuse and core plaques, DeMattos and colleagues found that in mE8-treated animals, the area covered by amyloid plaques stayed fairly steady, but their morphology changed, whereby plaques appeared mostly diffuse. In contrast, the BACE inhibitor-treated animals had almost no diffuse plaques, but core plaques were left behind. The combined therapies attacked both types. To DeMattos, these results suggest that the two different therapies attack different forms of plaque, with the BACE inhibitor cutting off the supply of Aβ that feeds formation of diffuse plaques and the mE8 antibody tackling plaque cores.
In a related poster, Margaret Racke, also from Lilly, detailed the Aβ fragments that predominate in the different types of plaque. Both diffuse and core plaques were made up mostly of Aβ40 and Aβ42, but the mixture of fragments they also contained differed. Probing western blots with antibodies the scientists developed to recognize different N-termini exposed by various secretases, they found that Aβ4-, 9-, and 11-42 were enriched in the diffuse plaques left over from the mE8 therapy. Aβ3- and 8-42 predominated in core plaques remaining after treatment with the BACE inhibitor. DeMattos said that these results help further understand the Aβ deposition cascade and give insight into the maturation of a plaque from diffuse to the more toxic core type.
Because the behavior of PDAPP mice is highly variable at 18 months of age, the scientists did not analyze it, DeMattos said. Instead, they analyzed dystrophic neurites and dendrites as a proxy for neuronal damage. These swollen neuronal extensions surround core plaques. They were prominent in untreated PDAPP mouse brains but appeared more normal upon amyloid removal with a monotherapy, even more so with the combination.
To confirm these findings, the scientists repeated the entire study with 19-month-old mice and got similar results. This time, the synergistic effect of the combination therapy reached statistical significance, DeMattos noted.
Researchers did not voice skepticism at the lack of behavioral data, noting that improved mouse behavior has not translated to clinical success in the past anyway. For commentators, the data on dystrophic neurites was sufficient to suggest a benefit on the central nervous system. Gunnar Gouras, Lund University, Sweden commented that the Lilly team might also have looked at synaptophysin, a synaptic protein that correlates with behavior.
“This is strong support for the notion of combination therapy,” said Dennis Selkoe, Brigham and Women’s Hospital, Boston. By attacking more than one form of Aβ, the combination is potentially lowering two sources of oligomers (monomers and mature plaques), which Selkoe and others believe are the most synaptotoxic species. He noted that a technically feasible addition to the study would be to examine what happens to soluble oligomer levels upon combination treatment. “Overall, I think this is the way to go in clinical trials, and we should start planning for them now,” he told Alzforum.
“The animal data [from this research and the Roche study] suggest we can more effectively lower amyloid using two complementary mechanisms,” agreed Reisa Sperling, also of Brigham and Women’s Hospital. A potential benefit of the combination approach is that a patient can take low doses of two medications, whereas a monotherapy might require a higher dose associated with more side effects. “Particularly in patients who likely have a significant amyloid burden, we’re going to need to be more aggressive with anti-amyloid therapy to reduce the buildup that’s already there,” she told Alzforum.—Gwyneth Dickey Zakaib
New efforts to tackle dementia are stirring to life around the globe, in part because U.K. Prime Minister David Cameron made it a prominent issue in his country’s 2013 presidency of the G8. Last December, he hosted leaders of the G8 (now G7) countries in London for the first summit dedicated to the illness (see Dec 2013 news story). From that, a World Dementia Council was appointed to stimulate innovation in treatment and care across the globe. The council includes 18 members from academia, industry, health care, and regulatory agencies. Following the summit, G7 countries planned four “legacy events” to address different issues in dementia care and research. At the Alzheimer’s Association International Conference 2014 held July 12-17 in Copenhagen, Denmark, the events’ hosts offered an update on their work.
“We feel the winds of change and collaboration, not only between researchers, but between countries, policy makers, and research funders,” said Yves Joanette, University of Montreal, Quebec, who chaired the session.
Dennis Gillings, World Dementia Envoy. [Image courtesy of OECD/Hervé Cortinat.]
On June 19, the United Kingdom hosted the first legacy event in London. Scientists, world leaders, policy makers, and funding organizations discussed barriers to investment in research and ways to stimulate it. Dennis Gillings, who was appointed World Dementia Envoy by Cameron, described how the U.K.’s Medical Research Council used the meeting to launch a £16 million public-private partnership called the U.K. Dementias Research Platform (see Q&A with Gillings below). The platform is intended to bring together data from two million research participants in 22 existing cohorts within the U.K., from healthy controls to people with early stage disease. International researchers will be able to access this diverse data from a central portal and extract epidemiological and biomarker data. Also at the meeting, Cambridge-based Alzheimer’s Research U.K. launched its Defeat Dementia campaign, which aims to raise £100 million for new research over the next five years.
Three more legacy events will take place between now and February 2015. On September 11-12, France and Canada will co-host a meeting in Ottawa that will focus on more closely linking academia and industry, using big data, and engaging the biotechnology and IT industries. During a three-day event in Tokyo on November 5-7, participants will learn about Japan’s patient-care initiatives and unique, community-based education. Finally, on February 9-10, 2015, stakeholders will meet at the National Institutes of Health in Bethesda, Maryland, to discuss how best to translate targets from basic science to clinical trials, new strategies to prevent disease, and new ways to assess and monitor it.
Beyond these official meetings, other countries and organizations have offered support, said Joanette. For instance, Scotland recently held a conference on strategies for better diagnosis and care, reducing crisis points for patients, and boosting participation in clinical trials. The World Health Organization (WHO) has pledged to assist in the exchange of knowledge between high- and low-income countries to share successful prevention and care strategies. The Organization for Economic Co-operation and Development (OECD) has pledged to help countries develop dementia plans. The OECD will host a big-data workshop in Toronto right after the Ottawa legacy event to examine what should be shared to accelerate research, as well as best practices for data sharing and barriers against it.
“The G7 legacy events indicate increased public awareness of dementia around the world,” said Ron Peterson, Mayo Clinic, Rochester Minnesota, who sits on the World Dementia Council. “There appears to be a solid commitment on the part of the G7 members to pursue strategies for dealing with dementia.”
At AAIC, some audience members wondered what will come of all of this. “What is the culmination of all this activity?” asked Zaven Khachaturian from the Campaign to Prevent Alzheimer’s Disease by 2020. “There need to be recommendations to OECD, the Alzheimer’s Association, WHO, or to various societies to translate your findings into actionable legislation.” Joanette responded that a concluding legacy event in March 2015 will assemble an action plan from reports of the preceding meetings. Others called for dementia to be added to the agenda of the G20 Summit in Australia, given that dementia extends beyond the developed countries of the G7.
Alzforum sat down with Gillings after the session. A biostatistician, businessman, and consultant to the pharmaceutical industry, Gillings is the founder of the Fortune 500 company Quintiles, the world’s largest biopharmaceutical development and commercial outsourcing firm.
Q: What is your role as World Dementia Envoy?
A: I’m trying to coordinate a variety of efforts throughout the world to accelerate disease modification. Research will get there in the end, but we don’t want to wait 20 or 30 more years. My role is to speed up the process.
Q: It seems a daunting task to essentially coordinate researchers, politicians, and industry around the world to conquer dementia.
A: It is, and one can’t coordinate everything that’s going on in the world. But what you can do is pick out things that need further stimulating, where there’s not enough effort, things that could be accelerated if you invested more. And you can also stimulate more collaboration. I think collaboration is one of the things that is not happening enough.
Q: How will the World Dementia Council accomplish these goals?
A: We’ve boiled everything down into three big priorities. One area is the regulatory environment. We want to change the risk/reward ratio. In dementia, 104 products have been put into clinical trials since 1993 and only three have been approved. We’d like to deploy progressive authorization so that applications for new drugs get a better consideration. I’ve also tried to put a special emphasis on dementia by coining the phrase “life-shattering disease.” It would be wonderful if regulatory agencies recognized this as a category; there might be an elevation of priority as a result of that designation. In terms of finance, we want to get more investment in research. The pharmaceutical industry is one big source, but we want governments to increase their investment. The third area I’m calling big data. We’d like a one-stop shop to access almost all data on dementia to facilitate more open-source collaboration in research, and more access to state-of-the art care for patients.
Q: What has changed since the G8 summit last December?
A: Awareness. It has increased by more than 50 percent, according to market research indicating that people over 50 in the U.K. are now more worried about dementia than cancer. That is amazing. It points to a shift in public perception. This galvanizes the politics. I would also say that David Cameron’s personal involvement has been enormously beneficial, because that automatically raises the profile.
Q: What’s the general feeling you perceive from politicians, scientists, and industry leaders facing the dementia challenge?
A: If I judge by these meetings and the legacy event in London, there’s increasing momentum and enthusiasm. With the U.S. setting out its plan in 2012, with the G8 formulating its strategy last December, and these legacy events being held, clearly things are happening. There’s also more funding. All that, I think, is elevating optimism considerably.
Q: Where will the necessary funds come from?
A: A variety of sources. First of all, governments are investing more. But the lion’s share, if this is going to be successful, will come from the pharmaceutical industry. That’s why the risk/reward ratio is critical. I would say that dementia needs at least a tripling of investment to have a hope of getting to our goal. That’s not going to happen overnight, but all this awareness, public perception, and media attention is good.
Q: Have any concrete initiatives or funding resulted from the G8 summit?
A: In the United Kingdom, there’s been an upping of government funding. Alzheimer’s Research U.K. announced their initiative to raise £100 million. David Cameron wrote to the other G7 countries about creating a global charter on dementia. My hope is that an international entity would come out of this, where my role and that of the World Dementia Council would evolve to have its own governance structure and financing, whereby these efforts get continued under a permanent executive leadership.
Q: What does this mean for the average person concerned about a loved one, or who has a family history of dementia?
A: If you’ve had dementia in your family and are concerned about genetic linkages, it would be nice to know we are making progress and that you won’t have to suffer from the same disease. If we can delay dementia onset by five years or more, that would be very substantial to the man on the street.
Q: Thank you for this conversation.
A: It’s important that researchers read and get energized about this.
See Gillings speak at the June 19 legacy event in London here.—Gwyneth Dickey Zakaib
Falling Dementia Rates in U.S., Europe Hint at Prevention Benefit
At the Alzheimer’s Association International Conference 2014, held July 12-17 in Copenhagen, Denmark, researchers presented more evidence that the risk of dementia is nudging downward in developed countries. The data add to a slew of recent studies that report similar findings (see May 2013 news story). According to results in a U.S. cohort, the incidence of dementia has declined gradually over the past 40 years. In Germany, a similar trend cropped up on a shorter time scale, and dementia appeared to occur over a shorter time period at the end of life.
As with previous reports, researchers attributed this reduction to improved cardiovascular risk factors and higher levels of education in certain countries. In absolute numbers, these incidence declines will be overwhelmed by increases in dementia brought on by population aging and negative health trends such as diabetes, but still, the data might shed light on means of delaying or preventing the disease, they said.
“Worldwide dementia cases will likely grow significantly over the next 40 years, but a number of studies now suggest that the declining risk of dementia in high-income countries has moderated that growth,” said Kenneth Langa, University of Michigan, Ann Arbor. He agreed that education and cardiovascular treatment could be responsible. “Going forward, it will be key to understand how these trends are working in low- and middle-income countries.”
Claudia Satizábal, from the lab of Sudha Seshadri at Boston University School of Medicine, presented new data from the Framingham Heart Study, a longitudinal cohort based in Massachusetts. The researchers became curious when they caught wind of falling dementia rates in other studies. To see if their population showed similar tendencies, they examined cognitively normal participants aged 60 and older starting in the late 1970s. From baseline assessments in four time windows of five years each, Satizábal and colleagues assessed how many people had progressed to dementia five years later. Relative to the first cohort assessed, the second, third, and fourth cohorts (in the '80s, '90s, and 2000s, respectively) had 22, 38, and 44 percent fewer incident cases of dementia. That decrease was stronger for women, and showed up more often in those with at least a high school diploma. Moreover, the mean age of dementia onset rose steadily, from 80 in the earliest time window to 85 in the last. Over the same time period, the researchers observed declines in stroke, blood pressure, cardiovascular disease, and smoking in the studied population.
The Framingham study examined mostly Caucasian people, but Satizábal said upcoming data from other studies will examine whether these trends hold in more ethnically diverse populations. “Dementia projections still hold and are worrisome,” she said at a press conference highlighting her work. “However, our results offer cautious hope that perhaps some dementia cases might be preventable by better management of cardiovascular risk factors and also by making education more available for all.”
Other researchers found much the same over a shorter time frame. Gabriele Doblhammer, German Center for Neurodegenerative Disease, Bonn, presented 2007-2009 data from AOK, the largest public health insurance provider in Germany. Using 2009 values as a reference and looking back in time, Doblhammer found that the prevalence of dementia in people aged 75-84 had been 2 and 4 percent higher in 2008 and 2007, respectively. This suggested a gradual decline in prevalence, the proportion of cases in a given age bracket each year. This effect, too, was strongest for women. The number of new dementia cases, i.e., the incidence, also dropped—2007 levels exceeded 2009 levels by 11 percent.
At the same time, mortality increased in people with dementia, especially women. That is actually good news, Doblhammer said, because it implies that new dementia cases are diagnosed at progressively older ages, and people spend less time with the disease. “This suggests that healthy life expectancy expanded, while the time with dementia became shorter,” said Doblhammer. She added that although the data contain no reasons for the shift, she also suspects education and cardiovascular risk factors.
Martin Prince, Kings College London, U.K., called the findings good news. “Incidence is happening later in life, closer to the time of natural death, therefore survival is shorter and we have compression of the time spent with dementia,” he said. However, trends look different in developing countries. Prince emphasized published data from China, where rising obesity, diabetes, and smoking is expected to compound huge increases in dementia brought on by shifting demographics (see Jun 2013 news story on Yang et al., 2013). “Population aging is happening much more rapidly in low- and middle-income countries,” he said. Therefore, health trends there will dominate global dementia levels.
Neil Buckholtz, National Institute on Aging, Bethesda, Maryland, pointed out that health gains in developed countries are likely to be undermined by rising rates of obesity and diabetes. “I’m not sure which trend will prevail,” he said. Population aging will still lead to many more cases of dementia, and they will be accompanied by a staggering price tag. “It’s hard to imagine how countries are going to deal with these costs,” Buckholtz said.—Gwyneth Dickey Zakaib
A nutritious meal plan, regular exercise, and other behaviors boost mental acuity, according to early results from a 1,260-person study reported at the Alzheimer’s Association International Conference in Copenhagen, Denmark, July 12-17. Miia Kivipelto of the Karolinska Institutet in Stockholm presented the first data out of the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability. The FINGER study is a randomized controlled trial that showed a multipart lifestyle intervention improved cognitive scores and, by inference, perhaps curtailed the risk of future Alzheimer’s disease. The large-scale trial stands poised to become the granddaddy of a coming wave of trials to tease out how lifestyle interventions affect dementia susceptibility. Several smaller studies are already ongoing, and researchers shared updates on four trials of exercise and health education programs in a separate symposium at AAIC.
“I was happy to see that in this conference there were many sessions on lifestyle interventions,” Kivipelto told Alzforum. “We need many more studies, we need to understand what is working for different at-risk populations.” In the past, epidemiological studies have indicated that healthy lifestyles protect against dementia. Researchers are only now starting to test that hypothesis directly in clinical trials.
Multi-FINGER Attack
Kivipelto thought it would be insufficient to modify just diet, or just exercise habits, or other single factors in the short term. Instead, the FINGER trial combined multiple interventions. The researchers recruited people aged 60-77 years who they thought could use some help. Specifically, Kivipelto invited people from previous population-based studies who scored high on a calculation of future dementia risk (Kivipelto et al., 2006). Cognitively, the participants performed at or slightly below average on neuropsychological tests.
The participants were randomized to one of two groups: an experimental cohort who received high-intensity interventions, and a control group who received moderate health advice. In that manner, the study was as double-blinded as is possible with lifestyle interventions; control participants still received some encouragement, and the researchers who rated outcomes did not know each subject’s assignment. Lon Schneider, a clinical trialist at the University of Southern California in Los Angeles, praised the study’s design. “This is a real experiment,” he told Alzforum. “The method of selection of patients and the randomization minimized biases.”
Specifically, people in both study arms received advice on how healthy eating, physical activity, mental stimulation, and social activities could abate vascular risk, which is linked to to one’s odds of developing dementia. They underwent blood pressure measurements and weigh-ins three times over the two-year study, and had four blood tests with written advice on their results.
People in the heavy intervention group underwent a much more rigorous regimen. They followed customized diet plans from a nutritionist. Under the guidance of a physiotherapist, they performed strength training and aerobic workouts. They exercised their brains with a computer-based cognitive training program. Several group meetings added social time. In addition, the participants attended more frequent meetings with nurses and physicians to monitor cardiovascular risk factors, such as blood pressure and weight, and received advice on improving those numbers.
The effort paid off, according to Kivipelto’s preliminary results. The two-year intervention period ended earlier this year, and the researchers have calculated their primary outcome measure, a composite score from the standard Neuropsychological Test Battery (NTB). Scores in the intervention group were about 40 percent higher than those in the control segment, Kivipelto said. Specifically, people in the arm with the intensive modifications performed better on tests of memory, executive function, and speed of thought. Kivipelto and colleagues still have plenty of data to analyze, but she told Alzforum that vascular risk factors declined among the intervention group, indicating that their likelihood of future dementia also shrank.
“The magnitude of that composite [NTB] outcome, although some people might say it is small, is certainly as large as what is expected with drugs,” Schneider said. “If this were a drug, the sponsors would be shouting, ‘Breakthrough!’”
Rachelle Doody of Baylor College of Medicine in Houston cautioned in an email to Alzforum that the NTB scores remained in the normal range. “This does not necessarily predict that the same intervention will delay or prevent the development of Alzheimer’s disease,” she wrote. Kivipelto and colleagues plan to follow their study participants for another five years to determine if there will be fewer incident cases of dementia in the intervention group. They will also analyze biomarkers such as Aβ from cerebrospinal fluid (CSF), as well as MRI and PET scans.
Delving into Details
The FINGER trial provides a proof of concept, Kivipelto said: “For sure, now we can say that lifestyle is important.” The study and its combined interventions furnish an ideal example for future work, commented Laura Baker of the Wake Forest School of Medicine in Winston-Salem, North Carolina. Also at AAIC, Baker and three other scientists showed progress reports on their own trials.
Baker has enrolled 50 people in her Piedmont Triad Aging, Cognition, and Exercise Study (PACE). These subjects are at high risk for future dementia thanks to both MCI and prediabetes, a condition in which blood sugar management is abnormal but not yet overtly diabetic. These folks are sedentary and truly need the fitness intervention, Baker told Alzforum. She is always concerned when she sees this MCI/prediabetes combination in her patients. “I know that when I see them in a year, they will have gotten worse,” she said.
The PACE participants undergo either an aerobic training program or a control stretching protocol. With 10 people still in the study phase, Baker said that interim results suggest improved executive function for the aerobic group. Baker is collaborating with Tom Montine at the University of Washington in Seattle to study CSF biomarkers. The people in the aerobic group exhibited reduced CSF tau and phosphorylated tau compared with baseline measurements. “Very few interventions lower tau,” Baker noted. On brain scans, the researchers noted increased blood flow in the anteromedial temporal lobe, a region that the Alzheimer’s Disease Neuroimaging Initiative has shown to be vulnerable in people who are likely to progress to AD.
Like Baker, Nicola Lautenschlager of the University of Melbourne in Australia is targeting sedentary people. With the Individual Goal Setting (INDIGO) trial, Lautenschlager aims to motivate them to exercise. Everyone in her study will receive a customized at-home walking program and regular phone check-ins from volunteer mentors. One motivating factor will be goals, set by the participants, such as being able to play with grandchildren without getting short of breath. Every subject will set goals, but those in the intervention group will regularly discuss their goals with their mentors. Lautenschlager has evaluated 14 of a planned 60 participants thus far. The main outcome will be how well participants keep up their activity, measured by personal records as well as pedometers and accelerometers.
Similarly, Linda Clare of Bangor University in the United Kingdom made goal-setting and mentorship key parts of her intervention. At AAIC, she described a trial within the AgeWell community center in Nefyn, Wales, targeted to people 50 or older. Seniors can partake in various activities such as art, choir, or dancing. In Clare’s trial, 75 participants were evenly split between those who only used the facility, those who also set individual goals in an interview, and those who set goals and received regular telephone mentoring. After a year, physical activity rose in both goal groups, but stayed the same in the controls. People who received mentoring did best.
A physical center like AgeWell may not be a good fit for everyone. Kaarin Jane Anstey of the Australian National University in Canberra is targeting tech-savvy elders with the online Body-Brain-Life program. This 12-week intervention includes seven educational modules on topics such as nutrition and dementia risk factors, and guided activities. Some participants received weekly face-to-face interactions in addition to the computer program. Control participants got regular emails about health topics. In this 176-person study, all groups improved their scores on a questionnaire about AD risk factors. The people who received online plus face-to-face interactions performed the best.
Lifestyle risk research was “left of field” when it emerged 10 years ago, Lautenschlager said, but its heavy presence in Copenhagen showed it has come of age. She and Kivipelto envision a future in which people at risk for dementia receive prescriptions for both medication and lifestyle changes to keep their brains healthy.—Amber Dance
Exosomes—small membrane-bound packets of cellular components that escape the cell—are coming under suspicion as players in neurodegenerative disease. Could they yield a biomarker? At the Alzheimer’s Association International Conference held July 12-17 in Copenhagen, Denmark, two researchers argued that exosomes found in blood hold the potential to signal disease. One looked at Alzheimer’s disease proteins packaged inside exosomes, the other examined the microRNAs therein. Both researchers reported ways to tell patients from healthy controls, and perhaps even predict future disease.
Multivesicular bodies (MVBs) fuse with the plasma membrane, releasing their inner vesicles as exosomes. [Image courtesy of Bellingham et al., 2012.]
Within the cell’s complex endosomal trafficking system, late endosomes called multivesicular bodies (MVBs) can either fuse with lysosomes for breakdown of their components, or merge with the plasma membrane and spill their intraluminal vesicles outside the cell (see image above). In the latter case, these intraluminal vesicles become known as exosomes. Containing tiny amounts of the cell’s cytoplasm, these packets essentially represent a sample of the intracellular environment, and are found in the cerebrospinal fluid (CSF), blood, saliva, urine, and milk. Previous studies have reported that exosomes contain proteins associated with neurodegenerative diseases, including Aβ, α-synuclein, and tau (see Rajendran et al., 2006; Emmanouilidou et al., 2010; and Saman et al., 2011). Given that exosomes have found some success as diagnostic biomarkers in cancer, some researchers wonder if they might also lead to a noninvasive biomarker of neurodegenerative disease (for a review, see Properzi et al., 2013).
Dimitrios Kapogiannis, National Institute on Aging, Bethesda, Maryland, generated excitement among attendees of an AAIC session when he presented new findings suggesting such a possibility for AD. He and colleagues isolated exosomes from the blood of 57 patients diagnosed with probable AD and 57 age- and sex-matched controls. The scientists enriched for exosomes from neural cells with antibodies for the neuronal adhesion proteins L1 and NCAM. While not expressed exclusively by neurons, these transmembrane proteins helped concentrate neuronal exosomes in the sample. The researchers then centrifuged the samples and used ELISA to quantify their levels of Aβ42, total tau (t-tau), and tau phosphorylated at T181 (ptau-181) or S396 (ptau-396).
Kapogiannis reported that the AD patients had significantly higher levels of Aβ42, ptau-181, and ptau-396 in their blood-based exosomes. Individually, the levels of each protein predicted AD with a percent accuracy in the high 90s. Combined, the three proteins’ predictive ability approached 100 percent, meaning they distinguished AD patients from controls with almost no overlap. T-tau yielded no difference between groups.
To see if the same approach could predict whether cognitively normal people would progress to dementia, Kapogiannis and colleagues sampled 24 patients who gave a blood sample at AD diagnosis and had already given another 1-10 years prior. These were matched with 24 more controls. All three proteins were elevated in early blood samples from those who later developed AD. While phosphorylated tau had already reached levels seen in diagnosed AD, Aβ42 measurements at these prediagnosis time points were intermediate between controls and patients, suggesting the peptide correlates with disease progression.
Together, the three proteins “near-perfectly discriminated between Alzheimer’s patients and controls,” Kapogiannis said. “There is also a possibility that these exosomal markers may … be used to predict disease at the preclinical stage.” Different proteins in exosomes could identify other neurodegenerative diseases, he pointed out.
Audience members lined up to ask questions after the presentation. One person wanted to know whether exosomes not enriched for neuronal origin gave a similar result. Kapogiannis said group differences were measurable even without selecting for neuronal markers, but were less distinct. Someone else asked whether the researchers planned to confirm these findings in exosomes from CSF, which would reflect neuronal sources even better. Kapogiannis responded that this had not been done yet because a high volume of CSF was required to obtain enough exosomes. A third listener asked about truncated tau in the exosomes, but Kapogiannis has not looked beyond those four proteins yet.
“That this test works in blood is encouraging,” said Reisa Sperling, Brigham and Women’s Hospital, Boston. She found the prognostic potential most intriguing, as it could make the test useful for the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s (A4) study to predict who has pathology. “To have a blood test that correlates with lumbar puncture results or PET imaging data would be an incredible breakthrough,” Sperling said, adding that it will be important to replicate the study in a larger sample of people characterized by biomarkers.
“The results were startling,” said Ralph Nixon, NYU Langone Medical Center, New York. “The degree of separation between AD and control and the ability to detect changes years before conversion was impressive.” He cautioned that the process of extracting exosomes and enriching for ones from neural cells is time-consuming for a technique that might be applied on a wide scale, but called the technical challenges surmountable. In the future, researchers might be able to refine the enriching marker to one that distinguishes neurons specifically, or even particular neuronal populations, he suggested. In addition, exosomal protein levels might hold clues to how disease disrupts the endosomal pathway, he said.
Researchers are also examining other exosome components as potential biomarker sources for Alzheimer’s. Andrew Hill, University of Melbourne, Australia, is examining the utility of microRNAs (miRNAs). These could be easier to detect than proteins because they can be amplified by PCR and do not require special antibodies, he said.
Hill and colleagues isolated exosomes from blood serum samples of 23 AD patients, three people with mild cognitive impairment (MCI), and 23 healthy controls from the Australian Imaging Biomarkers and Lifestyle (AIBL) study. Extracting and sequencing the RNA, the researchers identified 1,419 miRNAs, 220 of which showed up in all the samples. Of those, 14 were upregulated and three downregulated in AD and MCI compared with controls. A machine-learning technique called random forest modeling identified the 16 most predictive ones. They related to APP processing, apoptosis, and endoplasmic reticulum stress, Hill said.
To see if this set could predict disease, Hill’s group tested 15 more patients clinically diagnosed with AD as well as 35 healthy controls. All had undergone positron emission tomography with Pittsburgh Compound B (PiB-PET) to detect amyloid accumulation. Combined with information about age, sex, and ApoE4 status, the miRNA profile correctly diagnosed 13 of the 15 patients. It also correctly picked out 27 of 35 healthy controls, though five of the incorrectly characterized controls in fact had high amyloid burden. The researchers will wait and see if these five progress to AD in longitudinal assessments. If they do, it would mean the test has a specificity of 91 percent, he said.
Hill said he plans to examine a larger number of patients and validate the test in different cohorts, including more people with MCI. He will also check to see if the miRNA signature correlates with PiB-PET imaging. If so, the miRNA signature could become a low-cost, noninvasive screen for amyloid pathology. To extend the technique to other diseases, his group is looking at exosomes in people with Parkinson’s as well as animal models of other neurodegenerative disorders.
“This method has potential,” said Robert Nagele, Rowan University, Stratford, New Jersey, who chaired the session. The deep RNA sequencing makes it labor-intensive and expensive for now, but these are early days. Once the researchers settle on reliable biomarkers, they can streamline the method and develop simpler, cheaper assays, Nagele said. He noted that this method requires combination with clinical parameters to be highly accurate in a clinical situation. However, he said that for presymptomatic tests, it would be even better to find biomarkers that can stand on their own.
Exosomes appear to contain yet more proteins related to neurodegenerative disease. Markus Otto, University of Ulm, Germany, presented on a recent paper (see Feneberg et al., 2014), in which he and colleagues reported fragments of TDP-43 in exosomes from CSF. Quantifying these fragments in exosomes, or possibly phosphorylated and aggregated forms of the protein, may help diagnose TDP-43-related disorders such as amyotrophic lateral sclerosis and frontotemporal lobar degeneration in the future, he wrote to Alzforum in an email.—Gwyneth Dickey Zakaib
Denied Breakthroughs, Researchers Keep Hunting New Therapies
While Phase 2 results of crenezumab (see Part 2 of this series) grabbed headlines at this year's Alzheimer's Association International Conference, other unfolding stories warrant equal attention. Ana Graf from Novartis presented data from a Phase 2 dose-finding study for CAD106, an active immunotherapy against Aβ. This came the day after Novartis and Banner Health of Phoenix announced a partnership to test an active immunotherapy and a BACE inhibitor in cognitively normal adults who have two copies of the ApoE4 risk allele (see Part 1 of this series). Graf confirmed to Alzforum that this ApoE4 trial will test CAD106, but could not disclose the identity of the BACE inhibitor.
Novartis has been evaluating CAD106 in Alzheimer’s patients since 2005. The immunotherapy uses a carrier particle to present the N-terminal of Aβ (amino acids 1-6) to the immune system (see Aug 2006 conference news). The latest trial enrolled 121 people with mild AD to test the safety, tolerability, and immunogenicity of 150 or 450 mg of CAD106 injected with or without adjuvant. Secondary outcomes included amyloid PET, brain atrophy, serum Aβ, and CSF analysis of Aβ, total tau, and phospho-tau. Researchers gave 103 people up to seven injections of the immunotherapy over a period of 60 weeks, with final analysis at week 90. Fifteen people got placebo. This 7:1 randomization is unusual, said Graf, but it fit the study’s main safety and immunogenicity objectives.
Novartis monitored serological response, such as antibody titers. One person on the high dose and 15 on the low dose mounted no antibody response, and the company withdrew them from the trial. Overall, 54 percent on the low dose mounted a strong serological response, as did 76 percent on the high dose. About 8 percent of patients dropped out in the treatment group for safety reasons. Alum and MF59, the two adjuvants used, did not increase the antibody titers.
Graf said no major safety issues arose during the trial. The study detected amyloid-related imaging abnormalities, or ARIA, after dosing in five patients on CAD106, all of whom were strong serological responders. Four had ARIA-H, related to micro-hemorrhage, and one had ARIA-E, related to vasogenic edema. All five were asymptomatic; their ARIA was seen on MRI scans scheduled by the protocol. Even so, three of them were taken off the study because the protocol required a stop when at least two new micro-hemorrhages occurred, Graf wrote to Alzforum by email.
Serious adverse events related to CAD106 included dermatitis, atrial fibrillation, and acute psychosis. Adverse events occurred more often in patients on drug than placebo, but were not dose-dependent, said Graf. The use of alum seemed to minimize flu-like responses.
Plasma Aβ increased two- to fivefold in people who mounted a strong serological response compared to people on placebo, or to people on CAD106 who mounted no immune response. Amyloid PET imaging in 26 patients hinted that those with the highest antibody titers bound less florbetapir at 78 weeks than at baseline, and the correlation was statistically significant, said Graf. She said there were no changes in CSF Aβ, but CSF p-tau fell in those with the strongest antibody response.
On the small-molecule front, Ulf Neumann of Novartis presented a poster on the company’s BACE inhibitor NB-360.
Neumann told Alzforum that NB-360 differs from other BACE inhibitors in that it is based on a 3-amino-dihydro-oxazine core, rather than hydroxyethylene or ethanolamine. He said that while this core was difficult to synthesize, it may have advantages over the thiazine ring found in some other BACE inhibitors. The 3-amino-oxazine core allows for tweaks that render the compounds more brain permeable. NB-360 is highly selective over other aspartyl proteases, for example cathepsin D, and is potent in vivo in acute studies in mice, rats and dogs, according to the poster.
For a treatment study, Novartis tested the compound in APP51 mice, which express human wild-type amyloid precursor protein. The study used female mice beginning at 14½ months of age, when the animals have parenchymal Aβ deposits. Doses of 100 micromoles/kg of NB-360 for six weeks reduced Aβ staining in the brain, Neumann’s poster showed. Treated animals had fewer plaques, less Aβ40 and Aβ42 in the forebrain, and fewer activated microglia, as judged by Iba1 staining. The researchers did not record any behavioral changes. Neumann told Alzforum that proof of target engagement and a good toxicity profile are sufficient indications to develop the drug further.
These results are investigational, Neumann wrote to Alzforum; efficacy and safety have not been established for this compound, and next steps remain under review.—Tom Fagan
Scan by Scan, Growing Tau PET Data Picks Up Early Memory Deficits
This story has been modified from its original version.
Researchers have long known that tau pathology correlates more closely with the cognitive decline of Alzheimer’s disease than does Aβ pathology. They have predicted that being able to measure tau aggregates in the living brain would bring within reach a diagnostic and prognostic marker. At the Alzheimer's Association International Conference 2014, held July 12-17 in Copenhagen, Denmark, researchers reported that uptake of tau ligands into the brain as seen in positron emission tomography (PET) indeed correlates with memory decline. While this is good news, it is but a small step. "We are in the early days of tau PET and need to do much more work to be confident about what these ligands bind to in tissue. That said, tau imaging looks very promising," BradDickerson told Alzforum.
Dickerson, with Keith Johnson and colleagues at Massachusetts General Hospital in Boston and Charlestown, have performed some of the first clinical studies using the PET tau ligand T807, which is being developed by Eli Lilly. Johnson has examined binding of T807 in older adults who take part in the Harvard Aging Brain Study. This study tracks how approximately 250 cognitively normal people perform over time on challenging memory tests. It also peers into the brain using MRI and PET imaging of Aβ to see which pathological changes might precede or accompany changes in memory.
At AAIC, Johnson reported that among 56 people who underwent tau imaging, those who showed the greatest uptake of T807 in the inferior temporal lobe were also those who had deteriorated most in the previous three years on the selective reminding test. "Though preliminary, these findings suggest the spread of tau to the temporal neocortex signals memory decline in clinically normal older people," Johnson said. One implication is that tau imaging could be used for early diagnosis.
These emerging data fit with pathological staging of Alzheimer's disease described in the early 1990s (see Braak and Braak, 1991). Braak staging shows tau pathology spreading from the medial temporal lobe, mostly the hippocampus, into the cortex as the disease progresses. Interestingly, Johnson found that Aβ pathology also correlated with deterioration on the reminding test in this cohort of normal adults, but it did so independently of tau. The data suggest that tau spreads similarly in normal aging and in AD. "Basically, there is an age-related and an AD-related tau pathology, which share many common features," said Johnson. "The relationship between those two is of great interest."
Like Dickerson, Johnson emphasized that researchers still know too little about tau ligands and exactly what they bind to and where. Hints that certain ligands bind better to certain isoforms of tau, for example, raise the possibility of using specific tracers to diagnose specific diseases.
At AAIC, Makoto Higuchi from the National Institute of Radiological Sciences, Chiba, Japan, reported that the tau ligand PBB3 binds to tissue from Pick's disease patients, whose tau aggregates mostly comprise the protein’s three-repeat isoform. Other tauopathies, such as corticobasal degeneration and progressive supranuclear palsy, contain only four-repeat tau, while AD contains a mix.
Victor Villemagne, Austin Health, Melbourne, Australia, presented on THK-5117, a third tau ligand. While THK-5117 binds to PHF and the 4R lesions in PSP and CBD, it does not bind in Pick's disease. Then again, Higuchi reported that PBB3 does not bind to tau aggregates in hippocampal tissue from PS19 mice, which carry the human P301S tau mutation, yet it does bind to tau in the hippocampus of the rTG4510 model, which carries the P301L form. Interestingly, PBB3 bound to brainstem tau in PS19 animals. Higuchi suggested that there might be various toxic forms of tau that bind differently to the current crop of PET ligands.
"There is so much variability from case to case, and so many potentially confounding factors, that it will take a lot of effort to figure this out," Johnson agreed. He predicted that insight will come from postmortem correlations of tau pathology with prior PET scans taken when the study volunteer was alive. To date, researchers in the field have relied mostly on studying postmortem tissue using radiolabelled tau tracers, and in vivo studies are still gearing up.
As is the case with T807, researchers are getting a better handle on the properties and characteristics of PBB3 and THK-5117. In Copenhagen, Villemagne reported that THK-5117 has better binding kinetics than its forerunner, THK-5105. THK-5117 also seems to bind with greater specificity, since about 60 percent more gets taken up in AD brains than controls. That’s about twice as much as for THK-5105, which has but a 1.3-fold difference between cases and controls, said Villemagne. In 14 patients scanned with THK-5117 thus far, he observed higher binding in those whose MMSE scores were lower. Retention also correlated with loss of hippocampal volume. That correlation emerged in controls as well, though it was less robust.
As for PBB3, the number of patients scanned with this ligand has by now increased to 28—10 healthy controls, five people with MCI, and 13 with AD. As with THK-5117, binding of PBB3 correlated with MMSE scores, Higuchi said. Stay tuned on this fast-evolving topic.—Tom Fagan
Searching for Evidence of Neurodegeneration in the Blood
Given the invasiveness and expense of measuring brain biomarkers for neurodegenerative diseases, researchers are searching for alternatives. Despite little success thus far, they continue to comb the complex jumble of proteins and molecules in the blood (see Aug 2014 conference story and May 2014 conference story). At the Alzheimer’s Association International Conference 2014, held July 12-17 in Copenhagen, Denmark, researchers presented their latest prospects, which ran the gamut from proteins and protein fragments to antibodies and aggregates.
Signs of Alzheimer’s disease
A previous study led by Samantha Burnham, Commonwealth Scientific and Industrial Research Organization (CSIRO), Perth, Australia, found that a panel incorporating five proteins in the blood predicted neocortical Aβ burden with a sensitivity and specificity of 80 and 82 percent, respectively, in the Australian Imaging, Biomarkers, and Lifestyle (AIBL) cohort (see Burnham et al., 2013). The panel included chemokine ligand 13, immunoglobulin M-1, pancreatic polypeptide (PPY), interleukin-17, and vascular cell adhesion protein (VCAM-1). At AAIC, Burnham presented follow-up research to suggest that this same panel predicted progression toward AD in 585 healthy controls and 49 people with mild cognitive impairment (MCI) followed over 54 months as part of the AIBL study.
Eleven percent of healthy controls with the amyloid-associated blood profile progressed to MCI or Alzheimer’s disease (AD), compared with 4 percent of controls without it. Eighty percent of MCI patients with this blood signature progressed to AD, compared with 29 percent of MCI patients without it. Those with the AD-like blood profile declined faster on tests of episodic memory, noted Burnham. “This suggests that a simple blood-based signature not only provides estimates of neocortical Aβ burden, but also identifies individuals at risk of progressing to AD at the prodromal and preclinical stages,” she wrote in an email to Alzforum. “With further validation we hope that this panel could provide a frontline screening tool for recruitment to clinical trials.”
Nicholas Ashton, working with Simon Lovestone from Kings College London, also used AIBL participants to look for blood evidence of neocortical amyloid. Rather than a panel of five markers, these researchers used an unbiased mass spectroscopy (MS) analysis to examine the serum of 40 people with high and 38 people with low amyloid burden as determined by PiB PET scans. They identified more than 4,500 unique peptides reflecting almost 1,000 isoforms of 380 different proteins. Of these, 69 isoforms correlated with positive PIB scans. Because MS tests are difficult to translate into routine clinical assays, Ashton narrowed these hits down to 17 proteins that might be suitable for ELISA testing. Three proteins significantly associated with amyloid burden on ELISA tests: α-2-macroglobulin, FHR-1, and Fibrinogen γ chain (FGG). Notably, those differed from the five proteins fingered by Burnham.
In a second cohort from the University of California, San Francisco, consisting of healthy controls and patients with MCI or AD—47 with high and 32 with low PiB uptake—only FGG predicted amyloid burden. The researchers reported at AAIC that together, age and blood FGG levels predicted whether a person had neocortical amyloid build-up with 59 percent sensitivity and 78 percent specificity. FGG is involved in inflammation and homeostasis and has previously been associated with amyloid plaques (see Liao et al., 2004). However, FGG was not among the 10 plasma biomarkers that this group recently reported to predict conversion from prodromal disease to AD (see Hye et al., 2014). The researchers will next validate FGG in a larger cohort, focusing on cognitively normal participants, said Ashton.
“For predicting who should get screened into clinical trials, these projects could be very useful,” said Sid O’Bryant, University of North Texas Health Science Center, Fort Worth. While both sensitivity and specificity are somewhat low, O’Bryant suggested that blood tests could help screen a potential clinical trial population for those most likely to have amyloid buildup. Researchers could then perform more expensive PET scans on only the people with a positive test, limiting the cost of trial recruitment.
Other researchers are looking at antibodies that might signal disease. Robert Nagele, Rowan University, Stratford, New Jersey, explained that when a neuron dies, it releases molecules into the cerebrospinal fluid (CSF) that make their way into the blood. To clear the debris, the immune system produces autoantibodies, some of which may be specific to particular neuronal populations, Nagele said. He detects these serum autoantibodies by capturing them on a microarray of almost 9,500 human proteins.
Previously, Nagele reported that a panel of 10 autoantibodies distinguished commercially supplied blood of mild to moderate Alzheimer’s patients from that of controls with 96 percent sensitivity and 92.5 percent specificity (see Aug 2011 news story). At AAIC, he talked about extending the method to an earlier stage of AD and to other diseases.
Nagele examined blood samples from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). His panel separated 50 MCI patients whose low CSF Aβ42 put them at high risk of progressing to Alzheimer’s dementia, from 111 controls with a sensitivity and specificity of 98 percent each. These 10 autoantibodies were different from those he reported previously to detect mild to moderate AD, suggesting that there may be specific antibody signatures for different stages of the disease, Nagele said. Interestingly, the panel did not include antibodies against the Aβ peptide. It included an inflammatory marker, two microtubule-associated proteins, a growth factor regulator, an apoptotic regulator, and five uncharacterized proteins. Nagele plans to validate his MCI test in additional ADNI samples, especially from participants who enrolled as controls and later developed AD. Their archived blood samples would help determine whether this test can detect disease in presymptomatic individuals, he said.
Dave Morgan, University of South Florida, Tampa, praised the approach because it involved no a priori hypothesis, but instead looked widely for a signal that distinguished healthy people from others with AD or Parkinson’s disease (see below). Morgan sees a scientific rationale for picking up different autoantibody signatures in people with disease; however, the tests will need to be validated in much larger cohorts.
Nagele developed a separate autoantibody biomarker panel to distinguish people with early stage Parkinson’s disease from healthy controls. It correctly identified 89.2 percent of patients, he reported at AAIC. For clinicians looking for signs of neurodegeneration in general, Nagele’s group also developed a 50-marker panel that detected factors common to AD, PD, and multiple sclerosis. It differentiated disease from control samples with an error rate of 10.5 percent. Nagele said such a test could eventually serve as a general screen for neuronal damage, but it, too, needs validation in larger cohorts.
“The error rates seem quite reasonable,” said Morgan. If a blood test picked out people at risk for pathology with even 20 percent error, it would cut down on the number of more definitive, but expensive and invasive, tests used in people who have no pathology, he said. That said, since clinical diagnoses are wrong about 20 percent of the time, Morgan cautioned that Nagele’s actual error rate may be higher than calculated.
Blood Signature of Neurodegeneration
Other researchers are also looking for signs of general neurodegeneration. Dilek Inekci, working with Kim Henriksen of Nordic Bioscience, Herlev, Denmark, is pursuing tests for tau, which aggregates in several neurodegenerative disorders. While intact tau is difficult to detect in the blood, its fragments cross the blood-brain barrier more easily and some scientists think they could be a marker of disease. Inekci explained in a talk that after a neuron enters apoptosis, caspase-3 cleaves at the aspartate-421 site of tau and generates a C-terminal fragment called tau-C.
The researchers generated a monoclonal antibody specific for this fragment, which did not react with the intact form of tau. They used the antibody to immunoprecipitate the fragment from the serum of two AD patients and two healthy controls, and identified the immunoprecipitation products by mass spectrometry. While none appeared in the serum of normal controls, the fragment turned up in samples from AD patients. Quantifying the protein levels using ELISA, AD patients had twice as much tau-C as healthy controls.
Using the same antibody, the researchers developed an ELISA to detect tau-C and tried it on blood samples of eight people with hypoxic brain injury due to cardiac arrest. Patients had 10-fold more of the fragment than healthy controls. The researchers then performed the ELISA on blood from 18 people with AD, 18 with another type of dementia, and 18 controls. AD patients showed a non-significant trend of higher tau-C levels compared to the other two groups. “We don’t expect the fragment to be Alzheimer’s-specific, since you can see it rise in other cases of neuronal damage,” said Inekci. “Rather, it’s a marker of neuronal damage.” Such a marker could be used to predict concussion outcomes, Henriksen said. Inekci presented a separate poster reporting that a higher amount of tau-C reflected poor results following stroke in 19 patients.
Damian Crowther, University of Cambridge, U.K., is looking for a way to detect neurodegeneration in the blood by exploiting the tendency of many disease-related proteins to seed aggregation of normal forms. He and colleagues have developed a microfluidic approach to detecting seeds in brain and serum. The method involves loading a serum sample, fluorescently labeled synthetic Aβ42 monomers, and molten agarose onto a 1-centimeter plastic chip containing 10-50,000 picoliter wells. After incubating for three hours, the agarose cools into a gel, and researchers wash away unaggregated monomer. Any clumps greater than 1,000 kilodaltons stay behind. The researchers then look to see if any of the wells contain a fluorescing bundle. That would indicate that a seed was present in that well, i.e., the serum.
Crowther and colleagues applied this technique to brain extracts from flies and mice. It detected seeds in Drosophila that express Aβ42, but not in control brains. Likewise, the assay detected Aβ seeds in CRND8 mice, which overexpress a mutant version of human APP and overproduce Aβ42. It found seeds in serum samples from mice that were seven months old, an age at which they are robustly accumulating plaques. Crowther said he plans to examine how the test works in human serum samples. “This could be a good screen for drug candidates that stop the seeding process,” he told Alzforum.
One audience member asked if this seeding test works with tau or α-synuclein monomers. Crowther has not tried that but said it might be possible to multiplex this assay, where differently colored fluorescent tags added to a single sample label different types of monomer.—Gwyneth Dickey Zakaib
Researchers continue to target tau in hopes of treating patients with Alzheimer’s or other tauopathies. At the Alzheimer’s Association International Conference 2014 held July 12-17 in Copenhagen, Denmark, scientists presented their latest attempts to take aim at misfolded forms of this microtubule-binding protein. Potential therapies included two active vaccines, an antibody, an inhibitor of the enzyme that removes sugar molecules from tau, and an anti-aggregating compound. Most scientists are planning, or have already started, tests in humans.
An Immunological Approach
Some groups are working to harness immunological proteins to tame tau. Maria Pihlgren, AC Immune, Lausanne, Switzerland, presented a poster about the company’s active, liposome-based vaccine. It comprises a synthetic tau peptide attached to a linker that is bound to a lipid bilayer (see Hickman et al., 2011, and Jun 2012 conference story). This group tested various synthetic fragments that contain known pathological phosphorylation sites on tau to see which would generate a vaccine that best slows the progression of tau pathology in mice.
Pihlgren and colleagues tested the various vaccines in wild-type or tau transgenic mice that carried either the V337M/R406W or P301L mutations. They gave each three injections of the liposomal p-tau vaccines over three months. The tau antigens included Y18, S404/S409, S202/T205, T212/S214, and S396/S404 phosphorylation motifs. At the end of the treatment period, the scientists used an ELISA to measure the antibody response. Vaccines that targeted S202/T205, T212/S214, and S396/S404 elicited the strongest responses, but only the one aimed at S396/S404—called ACI-35—reduced aggregated tau in mice that carried the P301L mutation.
In treated mice, the ACI-35 vaccine helped animals retain body weight and delayed the onset of abnormal motor control. They also lived longer, with three times as many untreated mice dying by day 90. Having completed safety and efficacy data in mice (see Theunis et al., 2013), Pihlgren and colleagues began a Phase 1 trial in AD patients in December 2013. This is the first p-tau specific vaccine to be tested in humans. Andrea Pfeifer, CEO of AC Immune, told Alzforum that the company hopes to release data on the trial by the end of this year.
Another active vaccine is under development by scientists led by Michal Novak at Axon Neuroscience, Bratislava, Slovak Republic. Novak maintains that tau is truncated before it is hyperphosphorylated, and claims this cleavage step is therefore the root cause of neurofibrillary tangles in AD (see Feb 2013 news story). His company’s vaccine, AADvac1, targets this truncated, misfolded, pathological form of tau. It consists of a synthetic peptide derived from a tau protein sequence, which is coupled to keyhole limpet hemocyanin, a large antigen commonly used as an immune adjuvant. Novak declined to reveal the exact epitope. He presented safety and efficacy data from animals and preliminary results from a Phase 1 trial.
AADvac1 stopped tau phosphorylation and reduced neurofibrillary tangles in transgenic rodent models of Alzheimer’s disease, Novak reported. He uses a transgenic rat model that expresses truncated human tau. In preclinical safety testing in wild-type rats, rabbits, and dogs, no adverse effects occurred with acute doses or repeat injections for up to 26 weeks, Novak said. A Phase 1 study began last year in Austria. Thirty patients were slated to receive either placebo or vaccine for three months. All will then have the option to receive vaccine in a three-month, open-label extension. The scientists will monitor safety with magnetic resonance brain imaging, clinical and neuropsychiatric observation, cognitive testing, electrocardiography, blood biochemistry, hematology, coagulation measurement, and urine analysis. They will continue to observe patients in a separate 18-month follow-up trial. The company is planning to initiate a 24-month Phase 2 trial in patients with mild AD in early 2015.
Paired helical filaments (blue) accumulate outside of neurons (red) when antibody is applied at the same time (see text below).
[Image courtesy of Erin Congdon and Einar Sigurdsson.]
“Active immunization is a valid approach but riskier than injecting antibodies,” said Einar Sigurdsson at New York University Langone Medical Center, who thought that as long as there are no safety issues, such therapy would have more widespread use than passive immunization as it is cheaper to make and requires fewer injections.
Taking the passive antibody approach, Erin Congdon of Sigurdsson’s lab presented new findings about the tau antibody 4E6G7 (see Jun 2011 conference story). Neurons take up this antibody by clathrin-dependent Fcγ receptor endocytosis, whereupon it helps clear tau (see Congdon et al., 2013). Congdon reported that the antibody prevents uptake of paired helical filaments (PHFs) of tau into neurons. She grew primary neurons from mice in culture and incubated them with PHFs. In some cells she added antibody simultaneously, and in others applied it 24 hours later. In the latter group, antibody entered the cell and bound to tau aggregates. In the former, PHFs remained outside the cell, where they associated with the antibody. The findings suggest that the antibody formed complexes with the PHFs that prevented their uptake, said Sigurdsson.
Tau is thought to spread from neuron to neuron (see Aug 2013 conference story). To see the effect on spread between cells, Congdon cultured neurons on a special plate divided into two parts by a barrier that allowed only axons to pass through. She bathed the neurons on one side with PHFs with or without antibody. The untreated cells took up the tau fragments, which spread via the axons to the neurons on the other side. However, if 4E6G7 was applied at the same time or even 24 hours later, fewer PHFs spread to the neurons at the other end of the plate. “We think that the antibodies are acting intracellularly and extracellularly,” Sigurdsson told Alzforum, saying that multiple mechanisms are at play to prevent the spread of tau from cell to cell.
Disrupting Aggregation
Other scientists are working on therapies that prevent or break up the clumping of tau. Dirk Beher, Asceneuron, Lausanne, Switzerland, talked about ASN-561, his company’s inhibitor of O-GlcNAcase. This glycoside hydrolase removes sugars from tau and similarly modified proteins. Other groups also have adopted this strategy (see Mar 2012 news story). ASN-561 leaves more sugars on tau and makes it less likely to aggregate, reducing neurofibrillary tangles, explained Beher.
Because tau O-GlcNAcylation, or the process of adding sugars, decreases both with age and in Alzheimer’s disease, researchers believe that preventing the removal of sugars may restore tau's coat to a more youthful appearance (see Liu et al., 2009). Previously, they found that the inhibitor Thiamet G reduced O-GlcNAcase activity and prevented tangles from forming in tau transgenic mice (see Yuzwa et al., 2012). However, high doses were needed since the compound enters the brain poorly, said Beher. Asceneuron screened for an inhibitor that better entered the CNS, he said. After optimizing one of the hits, the group developed ASN-561, which potently inhibits O-GlcNAcase and crosses the blood-brain barrier.
To examine how it affected tau buildup, the researchers treated JNPL3 mice with ASN-561 for five days. They monitored O-GlcNAcylated tau with an antibody that specifically recognizes the O-GlcNAcylated form of the protein (see Cameron et al., 2013). In the forebrain, brain stem, and spinal cord of treated mice, the levels of O-GlcNAcylated tau rose dose-dependently up to 12-fold higher than they did in vehicle-treated mice, suggesting the compound hits the right target. So far, scientists have observed no safety risks with the compound, Beher said. Studies of tau aggregation and behavior are next.
If preclinical development is successful, the group plans to treat people with progressive supranuclear palsy, a rare, fast-progressing tauopathy. The hope is that PSP might lead to faster approval than if they started with Alzheimer’s disease. Beher said they are on track to begin the first human studies in late 2015.
Audience members asked about improvements in mice that express wild-type tau. Beher responded that he has not evaluated the compound in wild-type animals, given that they produce no neurofibrillary tangles. Others wondered if O-GlcNAcase knockouts are viable. Knockouts die before birth, Beher said. Pharmacological inhibition seems less drastic since Thiamet G had no major effects when given to mice for up to six months, he said. “We are not trying to inhibit the enzyme fully, just change the balance of O-GlcNAcylation to a healthier state,” said Beher.
While this is an interesting approach and the compound looks promising, these are still early days, said Eric Karran, Alzheimer’s Research UK, Cambridge. He commented that this is an example of an enzyme that has few known homologues and yet inhibiting it seems to have few toxic effects. “So far it looks like the body can tolerate ASN-561 pretty well,” he told Alzforum. Long-term toxicology tests will further explore that possibility. The group will also need to demonstrate that the compound attenuates tau pathology, Karran said. So far, scientists have struggled to tackle tau toxicity. “When it comes to affecting tau, there are not that many mechanisms that you can go after, so the more diversity of tau targets we have, the better.”
Jens Wagner, now at the German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany, presented data on a general oligomer buster called anle138b. Wagner formerly worked with Armin Giese, Ludwig-Maximilians-University Munich. Giese’s group previously reported that anle138b guards against prion and α-synuclein oligomer accumulation in mouse models of prion and Parkinson’s diseases, respectively, and prevents neurodegeneration and disease progression (see April 2011 conference story and Wagner et al., 2013).
To see if anle138b works in animal models of tauopathy, Wagner and colleagues gave the compound to tau transgenic mice. These animals overexpress human P301S tau, deposit the hyperphosphorylated protein in the cerebral cortex, hippocampus, brain stem, and spinal cord, and lose synapses and neurons, especially in the spinal cord, as they age. Treatment starting at weaning reduced the number of tau aggregates that formed before the animals died, according to Western blots. It also partially lowered synapse and neuron loss in the CA3 region and in the stratum lucidum of the hippocampus, according to stains for NeuN and synaptophysin. Treated mice performed better than controls in a novel object recognition learning and memory task. They also lived slightly longer—about two months—than untreated animals, though both survived significantly fewer months than wild types.
Giese and colleagues founded the company MODAG GmbH, which has licensed anle138b for further development. “Our hope is that next year we can start clinical studies,” said Giese. After preclinical safety testing, the group will start Phase 1 trials in healthy volunteers, and, if all goes well, Giese said they will move to Phase 2 in a population of people with a rare disease such as multiple system atrophy, a rapidly progressing synucleinopathy. These patients deteriorate quickly, which would allow scientists to measure outcomes in a relatively short timespan.
“Wagner provided extensive histological, neuropathological, and behavioral data for P301S mice treated with this drug,” said Eugenia Trushina, Mayo Clinic, Rochester, Minnesota, who chaired the session. “In combination with published work, I think they have solid data to support their therapeutic strategy.” Co-chair Thomas Wisniewski, New York University Langone Medical Center, agreed. “Aggregation is a nice target and he presented good data,” he told Alzforum. However, Wisniewski cautioned that the scientists started treatment in these mice before any misfolded tau had accumulated, so this represented a preventive strategy. He wondered if it would work as well after tau tangles had accumulated. By the time people with tauopathies are symptomatic, they have substantial tau buildup already, he pointed out. He also noted that the potential toxicity of anle138b compounds is unknown.—Gwyneth Dickey Zakaib

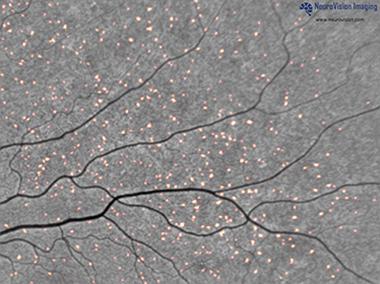
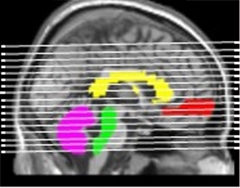

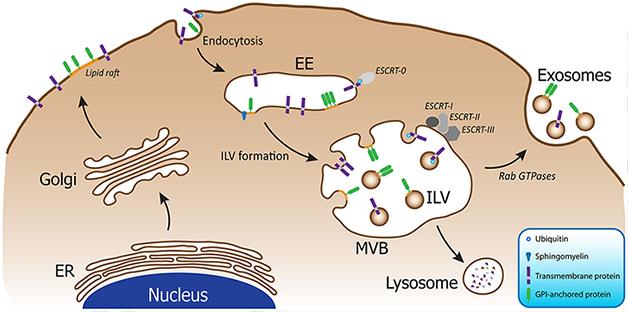
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.