Two Classes of Aβ Oligomers Act Differently in the Brain
Quick Links
Researchers know that soluble Aβ aggregates come in many shapes and sizes, but they have had trouble assigning specific effects to particular species in vivo and replicating those effects independently. In the June 4 Cell Reports online, researchers led by Karen Ashe at the University of Minnesota, Minneapolis, make another attempt. They distinguish between two broad classes of Aβ oligomers, reporting that type 1 and type 2 have distinct structures and behaviors. In mouse models, type 1 oligomers appear early, disperse widely through brain tissue, and correlate with subtle cognitive deficits. Type 2, on the other hand, have the structure of amyloid fibrils, appear only after plaques form, remain sequestered around plaques—and seem harmless to the mice. It is unclear if these data reflect what happens in human brains. Still, the findings highlight the importance of defining the specific oligomers present in Alzheimer’s, the authors suggest. This knowledge, coupled with information about antibody binding characteristics, might explain why some antibody therapies have worked better than others.
“I’m thrilled with this report. This is the clearest data yet showing that different oligomers have different pathological consequences,” said Charles Glabe at the University of California, Irvine.
Glabe was not involved in the research, but in previous studies he catalogued two general varieties of Aβ oligomer: those that have a parallel β-sheet structure similar to fibrils and bind the OC antibody, and those that have a distinct “prefibrillar” structure and bind the A11 antibody (see Kayed et al., 2007; Glabe, 2008). Later studies suggested that A11 recognizes β-sheets with an antiparallel, out-of-register conformation, in which every other strand travels in the reverse direction and amino acids do not line up (see Mar 2012 news; Liu et al., 2012).
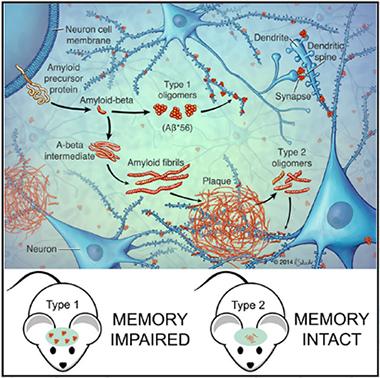
Two Pathways to Aggregation.
Aβ peptides form either small, type 1 oligomers, which stray into synapses and disrupt memory, or large, stable aggregates, which clump into plaques. Plaques then give rise to local, fibril-like type 2 oligomers. [Image courtesy of Cell Reports, Liu et al.]
Ashe wondered if these two oligomer classes might behave differently in vivo as well. To characterize them, first author Peng Liu prepared aqueous extracts from the brains of Tg2576 and hAPP-J20 mice of different ages. The former carry human APP with the Swedish mutation; the latter, Swedish and Indiana. For both models, the A11 antibody reacted with extracts from young and old mice, whereas OC only bound to samples from older, plaque-bearing animals. The authors then stained brain sections from Tg2576 (not the J20) mice with each antibody. OC lit up only dense-core plaques and their immediate surroundings, but A11 bound throughout brain tissue. This result highlights that the two types of oligomers appear at different times and places in the brain. The authors dubbed the A11 species type 1, and the OC variety type 2.
This study used A11 and OC binding to designate type 1 and 2 oligomers. It did not use biochemical or biophysical methods to characterize these species.
A third mouse model, rTg9191, expresses human APP with the Swedish and London mutations and develops plaques by 8 months of age (see Liu et al., 2015). In this model, type 2 oligomers again associated with plaques both temporally and spatially, but type 1 oligomers were absent at all ages. Because these mice express lower levels and a more amyloidogenic form of Aβ than the other models, the authors suspected that the expression level and amino acid sequence of APP might determine what kind of oligomers form. To test the idea that expression level matters, they used a fourth model, TetO-APPSweInd mice, in which transgene expression can be turned off. Switching off APP for five weeks in aged mice dropped type 1 oligomers by more than half, but type 2 oligomers remained stable. This confirmed that APP expression level affected each type differently, and that type 2 oligomers are more stable.
What effects do each type have on the brain? In earlier work, Ashe and colleagues traced memory deficits in Tg2576 mice to the presence of an A11-positive oligomeric species, Aβ*56 (see Mar 2006 news). Now classed as type 1, the presence of this 12-mer of Aβ correlated perfectly with cognitive problems in the other models as well, the authors found. The rTg9191 mice, which make no Aβ*56, performed a spatial memory or prefrontal behavioral task normally at any age. Likewise, turning off the transgene and thereby suppressing type 1 oligomers in TetO-APPSweInd mice has been reported to rescue cognition (see Fowler et al., 2014).
By contrast, type 2 oligomers caused no detectable deficits. Does this mean they are not toxic? To test this, the authors extracted type 2 oligomers from the brains of aged rTg9191 mice and injected them into the lateral ventricles of wild-type rats. The animals developed memory problems, forgetting a previously learned task. Type 2 oligomers can be toxic when dispersed through brain tissue, and are only innocuous in mice because they are sequestered around plaques, the authors concluded. Spatial distribution appears to be more important than quantity in determining toxicity, since type 2 oligomers make up about 95 percent of the total soluble Aβ in aged Tg2576 mice.
Overall, the data suggest a model in which Aβ can aggregate by one of two pathways, the authors propose. The peptide easily and quickly forms type 1 oligomers, but the 12-mers never grow into larger structures. Type 1 oligomers travel widely and therefore are most likely to interfere with synapses, leading to cognitive deficits. In the other pathway, Aβ more slowly forms parallel β-sheets that grow into fibrils and seed plaques. Once made, they persist. The plaque surface then catalyzes the formation of small, stable, type 2 oligomers with a parallel β-sheet structure similar to its own (see May 2013 news; Cohen et al., 2015). These oligomers remain confined to the vicinity of plaques, and therefore cause fewer cognitive problems. It is unclear what sequesters this species, though the authors speculate microglia may play a role.
Do these animal studies match what happens in people? Ashe and colleagues previously identified Aβ*56 in human brains, and reported that its levels rose with age, correlated with plaque load and pathological tau in early AD, but dwindled later in disease (see Lesné et al., 2013). In the present study, the authors measured plaque volume in postmortem AD brains, and found that plaque occupied 5 to 10 percent of the cortices. This is close to the plaque load in aged rTg9191 mice, which the authors calculated to cover 11 percent of the cortex. This suggests to Ashe that type 2 oligomers could be similarly restricted in human brains as in mice brains. Ashe said she will microdissect plaques and plaque-free regions from postmortem AD brains to test this question.
However, assuming that type 1 oligomers cause the cognitive decline associated with AD would be premature, Ashe said. She pointed out that the memory problems in mice are small, and might correspond to subtle cognitive deficits that crop up in preclinical stages of Alzheimer’s, not dementia. Dementia is characterized by massive neuronal loss, but neurons do not die in these mouse models. Perhaps there is some toxic ingredient missing in mice, such as human tau, Ashe suggested. In addition, plaques in human brains tend to form in the hubs of the default mode network, key nodes for neural communication (see Feb 2009 news). Thus, even if type 2 oligomers are confined to the vicinity of plaques, they might be able to disrupt this far-flung brain network, Ashe speculated. Connectivity in the DMN falters early in AD (see Nov 2007 news; Jul 2012 news). Further studies will be necessary to determine what kinds of cognitive problems each oligomer class causes in people, she added. Ashe also wants to nail down the fine molecular structure of the two classes, and determine which signaling pathways they activate.
Other researchers noted implications for antibody therapy. Lary Walker at Emory University, Atlanta, said future studies should determine how various therapeutic antibodies interact with different types of oligomers. This might explain why some antibodies work better than others. Ashe noted that bapineuzumab and gantenerumab both recognize fibrillar Aβ. In trials, these antibodies lowered plaque load but did not stem cognitive decline (see Sep 2012 conference news; Dec 2014 news). Aducanumab, which is reported to recognize Aβ oligomers, posted promising cognitive results in Phase 1 (see Mar 2015 conference news). Structural studies are beginning to decipher how various antibodies bind Aβ (see May 2015 news).
Distinct oligomeric species may also play a role in other protein-aggregation diseases. “This paper lays the groundwork for similar studies on other proteins implicated in neurodegenerative diseases, including α-synuclein, tau, TDP-43 and others,” Rakez Kayed at the University of Texas Medical Branch at Galveston wrote to Alzforum (see full comment below).—Madolyn Bowman Rogers
References
News Citations
- Anti-parallel Universe—Rare Amyloid Peptides in Cylinders, Sheets
- Aβ Star is Born? Memory Loss in APP Mice Blamed on Oligomer
- Aβ Fibrils Drive Oligomer Formation, New Model Suggests
- Cortical Hubs Found Capped With Amyloid
- Functional Imaging Gives Early Glimpse of AD
- Communication Breakdown: Multiple Networks Decline in AD Brains
- Bapineuzumab Phase 3: Target Engagement, But No Benefit
- End of the RoAD for Gantenerumab? Roche Declares Prodromal Alzheimer’s Trial Futile
- Biogen Antibody Buoyed by Phase 1 Data and Hungry Investors
- Shape of a Hug: How the Embrace of a Therapeutic Aβ Antibody Really Matters
Research Models Citations
Paper Citations
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. PubMed.
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008 Oct 31;283(44):29639-43. PubMed.
- Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A. 2012 Dec 18;109(51):20913-8. PubMed.
- Liu P, Paulson JB, Forster CL, Shapiro SL, Ashe KH, Zahs KR. Characterization of a Novel Mouse Model of Alzheimer's Disease--Amyloid Pathology and Unique β-Amyloid Oligomer Profile. PLoS One. 2015;10(5):e0126317. Epub 2015 May 6 PubMed.
- Fowler SW, Chiang AC, Savjani RR, Larson ME, Sherman MA, Schuler DR, Cirrito JR, Lesné SE, Jankowsky JL. Genetic modulation of soluble Aβ rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease. J Neurosci. 2014 Jun 4;34(23):7871-85. PubMed. Correction.
- Cohen SI, Arosio P, Presto J, Kurudenkandy FR, Biverstål H, Dolfe L, Dunning C, Yang X, Frohm B, Vendruscolo M, Johansson J, Dobson CM, Fisahn A, Knowles TP, Linse S. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat Struct Mol Biol. 2015 Mar;22(3):207-13. Epub 2015 Feb 16 PubMed.
- Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-β oligomers in ageing and Alzheimer's disease. Brain. 2013 May;136(Pt 5):1383-98. PubMed. Correction.
Further Reading
Primary Papers
- Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W, Shapiro SL, Reichl JH, Chiang AC, Jankowsky JL, Wilmot CM, Cleary JP, Zahs KR, Ashe KH. Quaternary Structure Defines a Large Class of Amyloid-β Oligomers Neutralized by Sequestration. Cell Rep. 2015 Jun 23;11(11):1760-71. Epub 2015 Jun 4 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UTMB
This is a nice piece of work and I commend the authors on their systematic analyses. From my own experience, I know how challenging working with oligomers and conformational antibodies can be. Amyloid formation in vitro and in vivo is very complex; thus, understanding the structural basis for amyloids is very important for basic biology and drug design. This is the first study that clearly reveals differences between two types of amyloid oligomers. Although we have known that antibodies A11 and OC recognize oligomers that are conformationally distinct (Kayed et al. 2007; Kayed et al., 2010), and follow distinct assembly pathways (Glabe 2008 and Krishnan et al., 2012), their distinguishing characteristics, formation in vivo, and toxicity were unknown.
The out-of-register oligomers recognized by A11 do not form parallel β-sheets found in Aβ fibrils in vivo. They are highly toxic in vitro and in vivo, and these dynamic structures exert toxicity via multiple mechanisms, such as synaptic dysfunction, pore formation, and tau aggregation.
Recently, we demonstrated that only A11-binding oligomers can cross-seed other amyloidogenic proteins. The results from this paper may explain this phenomenon and provide structural bases that can account for the formation of different amyloid oligomers in AD and other neurodegenerative diseases (Guerrero-Muñoz et al., 2014).
Important models for the out-of-register structures are available (Laganowsky et al., 2012, and Liu et al. 2012). This study suggests that therapeutic approaches, antibodies, and designed small molecules specifically targeting these structures may be beneficial for AD and other amyloid diseases.
Finally, this paper lays the groundwork for similar studies on other proteins implicated in neurodegenerative diseases, including α-synuclein, tau, TDP43, and others. Therefore, more methods and reagents will be needed in order to reveal the different amyloid structures.
References:
Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. PubMed.
Kayed R, Canto I, Breydo L, Rasool S, Lukacsovich T, Wu J, Albay R, Pensalfini A, Yeung S, Head E, Marsh JL, Glabe C. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol Neurodegener. 2010;5:57. PubMed.
Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008 Oct 31;283(44):29639-43. PubMed.
Krishnan R, Goodman JL, Mukhopadhyay S, Pacheco CD, Lemke EA, Deniz AA, Lindquist S. Conserved features of intermediates in amyloid assembly determine their benign or toxic states. Proc Natl Acad Sci U S A. 2012 Jul 10;109(28):11172-7. PubMed.
Guerrero-Muñoz MJ, Castillo-Carranza DL, Krishnamurthy S, Paulucci-Holthauzen AA, Sengupta U, Lasagna-Reeves CA, Ahmad Y, Jackson GR, Kayed R. Amyloid-β oligomers as a template for secondary amyloidosis in Alzheimer's disease. Neurobiol Dis. 2014 Nov;71:14-23. Epub 2014 Aug 15 PubMed.
Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic view of a toxic amyloid small oligomer. Science. 2012 Mar 9;335(6073):1228-31. PubMed.
Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A. 2012 Dec 18;109(51):20913-8. PubMed.
Federal University of Rio de Janeiro
Elucidating the identity(ies) of the specific Aβ assembly (assemblies) that is (are) most toxic to synapses and, hence, cause memory/cognitive dysfunction in AD remains a major research goal. Even though the role of Aβ in neuronal damage leading to dementia has been questioned recently (e.g., Herrup, 2015; Musiek and Holtzman, 2015), considerable evidence accumulated in the past 15 years indicates that soluble, diffusible Aβ oligomers are likely the proximal synaptotoxins in AD. However, the term “oligomer” constitutes a rather loose definition comprising assemblies ranging from dimers all the way to dodecamers and higher molecular mass species. Which of these oligomers is more closely associated with disease has baffled researchers and caused substantial controversy (Benilova et al., 2012).
Early studies in postmortem AD brain detected the presence of Aβ dodecamers (Gong et al., 2003), likely the same species subsequently termed Aβ*56 when detected in Tg mouse brains (Lesné et al., 2006) and verified in AD brains (Lesné et al., 2013). On the other hand, smaller assemblies (dimers/trimers) have also been found to accumulate in AD brains, to be potently toxic to neurons, and to disrupt memory (e.g., Klyubin et al., 2008; Shankar et al., 2008; Lesné et al., 2013).
This new study represents an important step toward solving the mystery of which Aβ species represents the most relevant target for diagnostics and therapeutics. By performing an extensive and careful analysis of the immunoreactivity of two different conformational antibodies—A11 and OC, which recognize different Aβ aggregates—in brain tissue from four different transgenic mouse models of AD, the authors have identified soluble and freely diffusible Aβ oligomers, which they termed type 1 AβOs, as the species associated with cognitive deficits in mice. Interestingly, they found that accumulation of type 2 AβOs, which are spatially restricted to the immediate vicinity of dense core amyloid plaques, does not affect memory in one transgenic mouse model investigated.
Further investigation of type 1 AβOs indicated that they comprise Aβ*56 and, possibly, other higher molecular mass species. This is an interesting result in light of other literature findings. We performed intracerebroventricular (icv) injections of high- or low-molecular mass oligomers isolated by size-exclusion chromatography under native-like conditions, and found that low molecular mass oligomers (ranging in mass from the equivalent of dimers to tetramers) caused loss of synaptophysin immunoreactivity and persistent memory impairment in mice (Figueiredo et al., 2013). On the other hand, high molecular mass oligomers (ranging from about 50 to 150 kDa) did not affect synaptophysin immunoreactivity and caused transient memory impairment, which was fully recovered by two weeks after icv injection. Interestingly, another recent study (Baker-Nigh et al., 2015) reported that high (but not low) molecular mass oligomers accumulate in AD forebrain, and may be involved in cholinergic dysfunction in AD.
An intriguing question arising from the combined results of Liu et al. (2015) and our previous findings (Figueiredo et al., 2013) is whether high molecular mass oligomers (i.e., Aβ*56 and higher) cause permanent or transient memory impairment in mice. Although this issue has not been directly addressed in the study by Liu et al., the finding that accumulation of type 1 AβOs is associated with progressive, age-dependent memory deficits in transgenic mice would argue that such oligomers cause persistent memory damage. Our previous results, by contrast, indicated that high molecular mass oligomers do not induce structural damage to synapses or persistent cognitive impairment.
The answer to this apparent contradiction may lie in the experimental paradigms used in the two studies. We used icv injections of synthetic Aβ oligomers into the brains of wild-type Swiss mice, whereas Liu et al. have examined the presence of oligomers in the brains of transgenic mice that express human APP variants leading to elevated Aβ levels. It is thus possible that, even though the impact of Aβ*56 and other high molecular mass oligomers might be transient, their constant production in the brains of Tg mice leads to persistently elevated levels, whereas injected oligomers may be cleared and/or detoxified in the brains of wild-type mice after a few days.
Yet another issue is the seemingly differential sensitivity of different brain regions to oligomer toxicity. As pointed out by Liu et al., one possibility is that the rate of activity-dependent Aβ production varies in different brain regions, thus favoring kinetic pathways leading to either type 1 (more neurotoxic) or type 2 (more benign) AβOs. Another possibility that remains open, however, is that different brains regions differentially express proteins involved in AβO binding to neurons and/or in the toxic signaling triggered by oligomers. In support of the latter possibility, we recently found that Aβ oligomers injected into the lateral ventricles of cynomolgus monkeys accumulated differently in distinct brains regions (Forny-Germano et al., 2014), in a pattern not clearly related to the distance from the site of injection.
It would be interesting to investigate the quaternary structures of type 1 and type 2 AβOs using biophysical/biochemical techniques, to complement the antibody-reactivity results of Liu et al. Although this appears a challenging task, it may pay off in terms of improved understanding of the roles of distinct Aβ assemblies and toward development of effective diagnostics and therapeutics for AD.
References:
Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015 Jun;18(6):794-9. PubMed.
Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and 'wingmen'. Nat Neurosci. 2015 Jun;18(6):800-6. PubMed.
Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012 Jan 29;15(3):349-57. PubMed.
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003 Sep 2;100(18):10417-22. PubMed.
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006 Mar 16;440(7082):352-7. PubMed. RETRACTED
Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-β oligomers in ageing and Alzheimer's disease. Brain. 2013 May;136(Pt 5):1383-98. PubMed. Correction.
Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008 Apr 16;28(16):4231-7. PubMed.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008 Aug;14(8):837-42. PubMed.
Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W, Shapiro SL, Reichl JH, Chiang AC, Jankowsky JL, Wilmot CM, Cleary JP, Zahs KR, Ashe KH. Quaternary Structure Defines a Large Class of Amyloid-β Oligomers Neutralized by Sequestration. Cell Rep. 2015 Jun 23;11(11):1760-71. Epub 2015 Jun 4 PubMed.
Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST. Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. 2013 Jun 5;33(23):9626-34. PubMed.
Baker-Nigh A, Vahedi S, Davis EG, Weintraub S, Bigio EH, Klein WL, Geula C. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer's disease. Brain. 2015 Jun;138(Pt 6):1722-37. Epub 2015 Mar 1 PubMed.
Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, Martinez AM, Klein WL, Houzel JC, Ferreira ST, Munoz DP, De Felice FG. Alzheimer's disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J Neurosci. 2014 Oct 8;34(41):13629-43. PubMed. Correction.
N. Bud Grossman Center for Memory Research and Care at the University Of Minnesota
University of Minnesota Medical School
University of Minnesota
Dr. Ferreira provides a nice summary of our study, links it to several interesting findings from his group, and offers his thoughtful suggestions on future directions.
First, we would like to point out that the concept of type 1 and type 2 Aβ oligomers (Aβo) is defined by their spatial, temporal, and structural relationships, with Aβ fibrils constituting dense-core plaques. The molecular size of type 1 and type 2 Aβo has been unclear yet. To our knowledge, research on the size of brain-derived type 1 Aβo is quite limited thus far besides that the conformation-sensitive A11 antibodies detect Aβo in brains of transgenic APP mice and AD patients of no smaller than hexamers (~25 kDa) under denatured western blot conditions (Lesne et al., 2006; Lesne et al., 2013). Unmasking the complete gamut of structural epitopes A11 antibodies recognize is critical to unveil the structural features, including the size, of type 1 Aβo. Out-of-register, anti-parallel-β sheet is one such epitope (Laganowsky et al., 2012; Liu et al., 2012), but it is highly likely there are others.
It is an interesting finding that Aβo influence cognitive function in a size-dependent manner. Though the size of type 1 and type 2 Aβo is not fully understood yet, a couple of observations from previous reports and this paper seem to support the findings of Dr. Ferreira and colleagues: 1) Aβ*56, a oligomeric assembly belonging to the type 1 Aβo, causes a transient spatial reference memory decline in healthy rats, when purified and injected into the animals (Lesne et al., 2006); 2) type 2 Aβo, showing their sizes no smaller than a 60 kDa globular protein in non-denatured size exclusion chromatography, lead to a transient work memory impairment in healthy rats, when type 2 Aβo-containing brain extracts were applied to animal brains (Liu et al., 2015).
Nonetheless, one of the key points we would like to deliver from this paper is that, in situ, spatial distribution is a key factor in determining the role of Aβo in neurological function. Should the sequestration of type 2 Aβo around plaques be disrupted, the benign Aβo would turn toxic, though in a different species. The distinct ex situ and in situ effects of type 2 Aβo add another layer of complexity to oligomer study, and suggest the importance and necessity of understanding the in situ effects of Aβo in future investigation.
What remains unsolved, however, is the spatial distribution of Aβo in AD brain. Do the findings in transgenic mouse models reflect what occurs in AD? This is the question we are currently addressing.
Finally, we agree that it is important to dissect the quaternary structures of type 1 and type 2 Aβo. We are developing methods to isolate individual Aβo species from a highly heterogeneous brain environment while maintaining their native conformations. We must also bear in mind that certain oligomeric species may fail to be discretely isolated because of the heterogeneous and metastable nature of Aβo. Biochemical/biophysical analyses would have to be carried out on a spectrum of conformationally analogous species.
References:
Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic view of a toxic amyloid small oligomer. Science. 2012 Mar 9;335(6073):1228-31. PubMed.
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006 Mar 16;440(7082):352-7. PubMed. RETRACTED
Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-β oligomers in ageing and Alzheimer's disease. Brain. 2013 May;136(Pt 5):1383-98. PubMed. Correction.
Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A. 2012 Dec 18;109(51):20913-8. PubMed.
Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W, Shapiro SL, Reichl JH, Chiang AC, Jankowsky JL, Wilmot CM, Cleary JP, Zahs KR, Ashe KH. Quaternary Structure Defines a Large Class of Amyloid-β Oligomers Neutralized by Sequestration. Cell Rep. 2015 Jun 23;11(11):1760-71. Epub 2015 Jun 4 PubMed.
Make a Comment
To make a comment you must login or register.