TREM2 Mystery: Altered Microglia, No Effect on Plaques
Quick Links
Since the 2012 discovery that mutations in the microglial gene TREM2 increase Alzheimer’s risk by about as much the ApoE4 allele does, researchers have tried to figure out why (see Nov 2012 news story). TREM2 being a microglial receptor, one popular idea holds that the risk variant prevents these immune cells from gobbling up amyloid plaques. However, in the June 2 Molecular Neurodegeneration, researchers led by David Holtzman at Washington University in St. Louis show it’s not that simple. In AD model mice lacking one TREM2 allele, the authors saw fewer microglia around plaques, but no increase in amyloid deposition. TREM2 variants exert their harmful effects through an amyloid-independent mechanism, Holtzman told Alzforum.
Other researchers were intrigued by the finding. Rita Guerreiro, Dervis Salih, and John Hardy at University College London speculated that TREM2 variants interfere with microglial migration, proliferation, or survival. “This is an interesting study that merits further investigation and replication,” they wrote (see full comment below).
First author Jason Ulrich crossed APP/PS1 mice with TREM2 knockout mice. The knockout mice were generated by Washington University’s Marco Colonna, who first cloned the TREM2 gene in 2000 (see Bouchon et al., 2000). The APP/PS1/TREM2 offspring had only one working copy of TREM2. Since the pathologic TREM2 variant, R47H, is believed to render the protein nonfunctional, the heterozygous TREM2 mice should mimic the effects of the R47H variant, Holtzman said.
The authors looked for effects on microglia and plaques in 3-month-old animals. They saw a dramatic difference in microglia. Far fewer of these immune cells gathered around plaques in the heterozygous mice compared to controls. These microglia also had smaller cell bodies than the controls did. Overall, the immune cells covered about 40 percent less territory around plaques in the heterozygotes. Despite this, the number and size of amyloid deposits did not change. (See image below.)
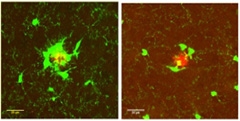
Fewer and smaller microglia (green) surround amyloid plaques (red) in TREM2 heterozygotes (right panel) compared to controls (left panel). [Image courtesy of David Holtzman and Jason Ulrich.]
Would a difference in plaques show up over time in older mice? To test this, the authors looked in 7-month-old heterozygotes. Again, they saw no change in amyloid deposition compared to controls. In ongoing work, the authors are examining whether these older mice also have fewer microglia around plaques.
Microglia can exacerbate Alzheimer’s disease by releasing cytokines that turn up brain inflammation. The authors measured levels of several cytokines to see if a lack of TREM2 might release the brakes on inflammatory processes. They found no significant differences between heterozygotes and controls, although a couple of pro-inflammatory cytokines were slightly decreased in the heterozygotes.
In future work, Holtzman will try to pin down what losing TREM2 in microglia does to the brain. Perhaps these cells have trouble phagocytosing molecules other than Aβ, or perhaps they interact differently with surrounding cells and intercellular matrix, Holtzman speculated. He also plans to examine the effects of TREM2 heterozygosity in other disease models, such as mice with mutant tau. Intriguingly, TREM2 variants increase risk for several neurodegenerative diseases that do not involve Aβ, including frontotemporal dementias (see Oct 2012 news story), Parkinson’s disease (see Oct 2013 news story), and amyotrophic lateral sclerosis (see Feb 2014 news story).
These mouse models also need to be tied to human disease. “Isolation of monocytes from TREM2 mutation carriers and analysis of their Aβ clearance and degradation functions will help to further delineate the involved mechanisms," suggested Michael Heneka at the University of Bonn, Germany (see full comment below). "Studies that combine such analysis with human PET imaging for Aβ of mutation carriers would be of great value,” he wrote.—Madolyn Bowman Rogers.
References
News Citations
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Mutations in TREM2 Cause Frontotemporal Dementia
- Fall Flurry of Letters Kicks Up Dust Around TREM2
- TREM2 Variant Doubles the Risk of ALS
Research Models Citations
Paper Citations
- Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000 May 15;164(10):4991-5. PubMed.
Further Reading
News
- Innate Immune Cells Enlisted to Clear Amyloid, Fight Disease
- Poor Digestion in Alzheimer’s Microglia: Is Beclin to Blame?
- Protective Microglial Gene Variant Promotes Phagocytosis
- Blessing or Curse? Peripheral Cytokines in the Brain
- Microglia Activation—Venusberg Meeting Questions M1, M2 Designations
- ApoE Disrupts Brain Networks, Helps Microglia Clear Aβ
- The Brain Minus Microglia—No Effect on Plaques
Primary Papers
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014 Jun 3;9:20. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Klinik und Poliklinik für Neurologie
Genetic variants in TREM2 have been found to increase the risk of developing Alzheimer´s disease. This paper by Ullrich and colleagues aims to identify molecular mechanisms underlying this risk. Previously, TREM2 expression has been reported in microglia, the brain´s major innate immune cell type. Since TREM2 was shown to be involved in the regulation of clearance of neuronal debris, these investigators hypothesized that TREM2 may be involved in phagocytic Aβ removal. While they found no changes in Aβ deposition in hemizygous TREM2-deficient mice, the number of plaque-associated microglia was reduced.
These results may be explained by various factors. First, TREM2 could be a specific receptor for the phagocytic removal of neuronal debris and not be involved in Aβ clearance per se. Another possibility is that toxic Aβ peptides affect the function of TREM2 in the mice they tested, thereby causing a functional impairment equal or similar to the hemizygous genetic knockdown. Third, a complete, homozygous knockdown of TREM2 may be required to influence Aβ levels.
Also, one may speculate that TREM2-mediated effects require binding of a specific, not yet identified ligand, which was not present at the time the animals were studied and may modulate TREM2 function at higher levels of Aβ deposition.
That TREM2 hemizygous deficiency reduced the number of plaque-associated microglia is very interesting. This clearly indicates that TREM2 is either involved in the microglial migration toward plaques, positively driving microglial proliferation at plaque sites or, in contrast, enhances microglial cell death in the presence of Aβ.
Overall, this paper makes the reader curious to learn more about TREM2 functions in AD and, in particular, about the effects of a homozygous genetic-deficiency model. Also, isolation of monocytes from TREM2 mutation carriers and analysis of their Aβ clearance and degradation functions will help to further delineate the involved mechanisms. Studies that combine such an analysis with human PET imaging for Aβ of mutation carriers would be of great value.
University of Southern California
In this interesting report, David Holtzman’s group has provided the first test of TREM2 heterozygous loss in a mouse model of cerebral amyloidosis (APPPS1-21). Their report is both timely and provocative, given recent GWAS results implicating a single allelic variant of TREM2 in increased risk for late-onset AD. Intriguingly, the authors found fewer plaque-associated microglia TREM2 heterozygous APP/PS1 mice, and remaining microglia were smaller in size and bore fewer processes. Yet, there was no significant change in Aβ deposition in this model. While this negative finding is surprising, it nonetheless agrees with results from Mathias Jucker’s group (Grathwohl et al., 2009), who demonstrated that spatially and temporally restricted deletion of microglia does not impact the nature or extent of Aβ deposits in APP23 transgenics. This, of course, leads one to wonder whether complete deletion of TREM2 would have a stronger impact on Aβ pathology.
References:
Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009 Nov;12(11):1361-3. PubMed.
Universitat Autònoma de Barcelona
This necessary study supports, once again, the idea that microglia do not affect plaque formation in mouse models unless they are "activated" by drugs or immunotherapy or replaced by fresh macrophages. Whether this applies to humans, too, remains to be determined.
Van Andel Institute
University College London
Institute of Neurology, UCL
We (Guerreiro et al., 2013) and others (Jonsson et al., 2013) have previously described a significant association between the TREM2 p.R47H variant and an increased risk for the development of Alzheimer's disease. Additionally, Trem2 expression was found to differ between control mice and a mouse model of Alzheimer's disease (TgCRND8 mice) when using an expression microarray assay. Expression of TREM2 was found to rise in parallel with a rise in cortical levels of beta amyloid.
These data suggested a role for TREM2 as a control gateway for microglial responses. In theory, a compromised function of TREM2 would likely affect the clearance of cell debris and, possibly, the removal of beta amyloid in Alzheimer's disease. At the same time, relief of the TREM2 lock on cytokine levels could fuel inflammatory cascades, leading to a systemic inflammatory response and the incidental death of neurons.
In order to shed some light on how TREM2 genetic variability acts on the pathological mechanisms observed in Alzheimer's disease, Ulrich and colleagues used APPPS1-21 mice, heterozygous for TREM2 (expressing only one allele of TREM2), following the hypothesis that the heterozygous variants described in AD induce a partial loss of function in the gene. The authors found no significant difference in Aβ deposition in 3-month old or 7-month old mice expressing one or two copies of TREM2, but identified a marked decrease in the number and size of plaque-associated microglia in 3-month old mice with one copy of TREM2. These data suggest that TREM2 is important for the microglial response to Aβ deposition but that a 50 percent decrease in TREM2 expression does not affect Aβ plaque burden.
This is an interesting study that merits further investigation and replication. The suggestion that microglia migration may be defective in Trem2 heterozygotes is of particular interest when taking into account our previous network analysis. That linked TREM2 to genes involved in cytoskeletal rearrangements—the basis of phagocytosis and migration (Forabosco et al. 2013). It would have been interesting to see results of a microarray analysis instead of specific microglial and inflammation markers. This data would allow the detection of any other crucial events, particularly if microglia are being pushed to a different non-M1/M2 state/phenotype.
Although the authors mention the possible codependency of TREM2 with CSF1R and the role of CD33 in microglia responses, given the previous literature on the genetics and biopathological function of these two proteins, they fail to further explain the trend for decreased levels of C1qa Ulrich and colleagues detected in TREM2 heterozygotes. C1qa is a component of the complement system which has also been strongly implicated by GWAS in the etiology of Alzheimer's disease.
In summary, the findings presented for this particular model of Alzheimer's disease indicate that a loss of a single TREM2 allele decreases the number and size of plaque-associated microglia in 3-month old mice, but has no effect on the total amyloid burden in either 3- or 7-month old mice. This suggests that the increase in the odds of developing AD caused by a possible partial loss of function TREM2 variant does not exert its effect through a defective clearance of amyloid plaques, but through another microglial function possibly associated with its activation, survival, proliferation, or most probably, migration.
References:
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):117-27. Epub 2012 Nov 14 PubMed.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):107-16. Epub 2012 Nov 14 PubMed.
Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, Weale ME, Ryten M. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 Dec;34(12):2699-714. PubMed.
View all comments by John HardyCase Western Reserve University
The mice analyzed by Ulrich et al. express only one copy of CX3CR1 (all mice, irrespective of TREM2 status, are CX3CR1+/GFP), and that could complicate interpretation of their findings. The effects, or lack thereof, of TREM2 deletion are superimposed on CX3CR1 hemizygosity. Although CX3CR1+/GFP microglia are thought to function normally in many cases, studies show that AD mice with one copy of CX3CR1 show significant differences from those with two copies (Lee et al., 2010). TREM2 has captured imagination of the field to such an extent that similar studies with wild-type microglia with complete loss of TREM2 function will likely appear soon.
The caveat of CX3CR1 hemizygosity notwithstanding, these are important findings and it is nice to watch the term "amyloid-independent mechanism" gaining a wider acceptance in AD discussions.
References:
Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010 Nov;177(5):2549-62. PubMed.
Certara
Our surprise reaction at this result might have been driven by our amyloid-centric view of Alzheimer’s disease. Effects on amyloid phagocytosis might not be the primary process driving vulnerability to certain TREM2 SNPs. TREM2 does other things besides amyloid phagocytosis and plays a role in other neurodegenerative diseases, such as models of stroke (Sieber et al., 2013) and its therapeutic hypothermic aftereffects, and in models of accelerated senescence, such as SAMP-8 mice (Jiang et al., 2014). TREM2 affects the release of cytokines and microglia migration. It might be a good time to start integrating various bits of information from adjacent fields of research to come up with a more comprehensive view of the many complex and non-linear interactions that play a role in the brain immune system.
References:
Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8(1):e52982. Epub 2013 Jan 3 PubMed.
Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, Tan L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014 Jun;35(6):1243-51. Epub 2013 Dec 2 PubMed.
Kawabori M, Hokari M, Zheng Z, Kim JY, Calosing C, Hsieh CL, Nakamura MC, Yenari MA. Triggering Receptor Expressed on Myeloid Cells-2 Correlates to Hypothermic Neuroprotection in Ischemic Stroke. Ther Hypothermia Temp Manag. 2013 Dec 1;3(4):189-198. PubMed.
Make a Comment
To make a comment you must login or register.