Fall Flurry of Letters Kicks Up Dust Around TREM2
Quick Links
Nearly one year ago, two research groups reported that a mutation in the triggering receptor expressed on myeloid cells 2, better known as TREM2, raises risk for Alzheimer’s disease by nearly as much as does the established risk factor ApoE4. Scientists around the globe started looking for the arginine-47-histidine variant in population samples and exploring how—and how robustly—the gene contributes to disease. In seven letters to the October 17 New England Journal of Medicine, researchers now debate the strength of the AD connection, add African descendants to the list of populations affected, suggest the mutation accelerates brain atrophy, and even hint that it contributes to Parkinson’s disease. In addition, the September 27 Journal of Biological Chemistry online carried news of an unexpected link between the core Alzheimer’s pathogenesis and the new gene, reporting that TREM2 relies on γ-secretase cleavage for proper signaling.
At Odds Over Odds Ratio
In the original reports, two research groups found that the rare R47H variant of TREM2 strongly associated with AD. Other variants were even rarer and more difficult to directly link with the disease (see Alzforum News story on Guerreiro et al., 2013 and Jonsson et al., 2013). Just how big a risk is having the R47H allele? The data suggested carriers had a three- to fourfold risk for AD, an odds ratio much higher than for any other AD gene except ApoE4. Now, writing in NEJM, Lars Bertram of the Max Planck Institute for Molecular Genetics in Berlin, Germany, and Antonio Parrado and Rudolph Tanzi of Massachusetts General Hospital in Charlestown take issue with that figure. The trio genotyped 6,421 DNA samples, mostly American in origin, from family-based datasets and case-control series from a variety of sources. Their meta-analysis suggested an odds ratio of only 1.7 for the R47H variant.
Since writing the letter, Bertram and colleagues added about 3,000 more subjects from European case-control cohorts, and the odds ratio dropped to 1.5, Tanzi told Alzforum. “The genetics are not as overwhelming as the original papers stated,” he said. Seeing an odds ratio drop after the initial publication should be no surprise, he said; original data often suffer a “winner’s curse” that inflates the significance of genetic associations (Ioannidis, 2008).
In an NEJM reply, Rita Guerreiro and John Hardy of University College London, authors of one of the original TREM2 reports, provided meta-analysis of data from the original two papers as well as more recent studies (Pottier et al., 2013, Benitez et al., 2013, Giraldo et al., 2013). They reported the odds ratios averaged 3.4 for R47H, though 1.7 frequently fell within the 95 percent confidence interval, indicating that statistically there is little difference between the new NEJM analysis and other studies.
The exact numbers will depend greatly on the particular sample surveyed, Guerreiro pointed out. “Subtle differences in population structure can influence odds ratios,” agreed Jordi Clarimón of the Sant Pau Hospital in Barcelona, Spain. “With rare variants, you can see this kind of incongruence.” In other words, even a couple of additional carriers in the case or control sample could change the final number for the odds ratio. Also writing in NEJM, Thorlakur Jonsson and Kari Stefansson of deCODE Genetics in Reykjavik, Iceland, the authors of the other original TREM2 paper, suggested that the size of the sample and the way the controls were selected could contribute to discrepancies.
Guerreiro and Hardy’s meta-analysis reflects the fact that studies from Utah, Spain, France and Belgium have confirmed the TREM2-AD link (Gonzalez Murcia et al., 2013, Cuyvers et al., 2013). In this week’s NEJM, researchers from the Alzheimer’s Disease Genetics Consortium add Americans of African descent to the list of populations affected. Based on a study of 1,968 cases and 3,928 controls, they report that several single-nucleotide polymorphisms in the TREM2 region are significantly associated with Alzheimer’s disease. In another recent paper in the September 13 Neurobiology of Aging online, Clarimón and the dementia genetic Spanish consortium confirmed links between the R47H variant and Alzheimer’s in people of Spanish descent, based on a study of 3,172 people with AD and 2,169 controls.
It will be important to continue examining TREM2 in various geographical locations, because the frequency of rare genetic variants such as R47H—found in fewer than 1 percent of people—tends to vary between different populations, said Clarimón. For that reason, he said, researchers must be careful to study large samples of homogeneous populations with well-matched controls, otherwise they risk reporting false associations, or missing real ones, due to biased samples (see full comment, below, and Tennessen et al., 2012).
Regardless of the true odds ratio, TREM2 represents a major risk factor for AD, with an effect size well above variants discovered in genome-wide association studies, said experts who spoke with Alzforum. “Whether [the odds ratio] is 1.5 or 1.7 or 3.0 is not so important, although I think it is closer to 3.0,” said Carlos Cruchaga of Washington University in St. Louis, Missouri. “The most important thing is that TREM2 is a new gene implicated in Alzheimer’s disease.”
“Bottom line is, [the R47H mutation] is a catastrophe … compared to the tiny risks the field has been studying,” added Paul Thompson of the University of Southern California in Los Angeles. “It is academic whether your risk is doubled or tripled … TREM2 is substantially more dangerous than any other gene, besides ApoE4.” Even an odds ratio of 1.7 would be high for a neurological disease, Thompson said.
The 2012 papers were not, in fact, the first time TREM2 and AD had been linked. As Thomas Bird, University of Washington in Seattle, noted in a letter to the NEJM, a hint arose in 1983 when he and colleagues found amyloid plaques and tangles in a member of a family with the bone disease Nasu-Hakola, which was later shown to be due to TREM2 mutations (Bird et al., 1983, Paloneva et al., 2002). “Because of that person’s young age, this was not likely to be a coincidence, and we suggested a possible relationship between Nasu-Hakola disease and Alzheimer’s disease,” Bird wrote. “Little did we know this relationship would be confirmed almost 30 years later.”
TREM2 Beyond Alzheimer’s
TREM2 mutations were linked to frontotemporal dementia (FTD) before Alzheimer’s, suggesting the gene could be a risk factor for neurodegeneration in general. Cruchaga and colleague Bruno Benitez tested this by looking for the R47H variant in people with Parkinson’s disease. They genotyped two separate cohorts: 478 people with PD and 837 healthy controls from a Washington University series, and 654 people with PD and 550 controls from the University of Navarra in Pamplona, Spain. They found three TREM2 mutation carriers with PD in the U.S. dataset, and six carriers with Parkinson’s in Spain, but no instances of the variant in any controls. Their findings echo those of Rosa Rademakers and colleagues at the Mayo Clinic in Jacksonville, Florida, who earlier this year reported that TREM2 R47H more than doubled the risk of PD in Caucasians of North American, Irish, and Polish ancestry (Rayaprolu et al., 2013).
No consensus yet exists on the link between TREM2 variants and PD, Cruchaga said. Guerreiro told Alzforum she checked her data for a PD link, and came up dry after examining more than 1,300 people with Parkinson’s. Jonsson and Stefansson, in their letter, also noted that they had found no significant association between TREM2 and PD in a study of 2,730 cases and 73,710 controls. The particular controls selected may explain the discrepancy, Guerreiro and Cruchaga agreed. Because the variant’s frequency in the population is so low, it may by chance be absent in some control groups but present in in another, arguing against a link with PD.
While R47H is the main TREM2 variant linked to Alzheimer’s, it remains to be seen which TREM2 mutations contribute to FTD. Guerreiro and Hardy found that mutations that caused early termination of the TREM2 mRNA led to FTD in Turkish families (see Alzforum News story on Guerreiro et al., 2012), and another group found a nonsense mutation in familial FTD in Colombia (Giraldo et al., 2013). Researchers debate whether R47H increases FTD risk, as well. The Mayo Clinic study earlier this year reported it did, with an odds ratio of 5.0, as did a recent study in Belgium which calculated an odds ratio of 6.2 for TREM2 variants and FTD. However, Clarimón’s group found no TREM2 mutations in 682 people with FTD in Spain, and other researchers found no instances of TREM2 R47H in 175 people with FTD in France (Lattante et al., 2013).
TREM2 on the Brain
Given the rarity of the R47H variant, experts told Alzforum it will be of little utility as a diagnostic or prognostic marker. Rather, it is important because it offers a biological clue to pathology. Already, Thompson’s group reported in NEJM a direct effect of TREM2 on neurodegeneration. First author Priya Rajagopalan and colleagues correlated TREM2 variants with magnetic resonance images, taken two years apart, of 478 people participating in the Alzheimer’s Disease Neuroimaging Initiative. The group included 100 people with AD, 221 with mild cognitive impairment, and 157 healthy controls. The sample contained seven TREM2 mutation carriers, all with AD, who suffered “a wildfire of tissue loss” in the brain, said Thompson.
On average, healthy elderly people lose about 1 percent of their temporal lobe tissue per year. People with AD lose about 3 percent. Carrying the TREM2 R47H variant doubled that rate to approximately 6 percent for carriers with Alzheimer’s, Thompson said (see image below). “You could not sustain that [loss] and have no problem,” he pointed out. Symptoms of Alzheimer’s emerge after about 10 percent of the medial lobe is lost, meaning that people with TREM2 mutations are likely to develop the disease years earlier than others, noted Thompson.
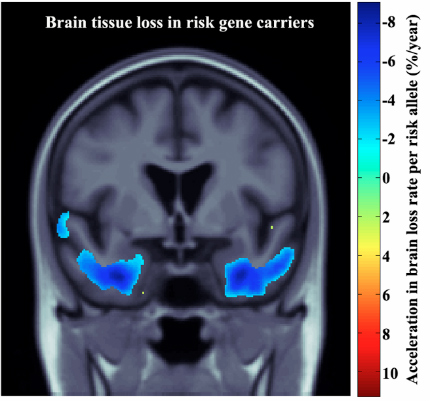
Brain drain: TREM2 mutation carriers lost 1–3 percent more of their temporal lobe tissue per year, compared to noncarriers, as shown in this compilation of NMR images from several study participants. Paul Thompson, University of Southern California, Los Angeles.
This study proves that the TREM2-risk allele damages the brain, and that TREM2 plays an important role in the organ’s maintenance, said Monica Carson of the University of California at Riverside, who was not involved in the research. Thompson suggested recruiting people with TREM2 mutations for clinical studies, because their faster decline could be easily measured. One could do a shorter trial, or include fewer participants, by enriching for TREM2 carriers, he said. Though the variant is rare, Thompson thought it would be possible to recruit 200 carriers.
TREM2 and APP Share a Secretase
Confirming links to PD and FTD would indicate a general role for TREM2 dysfunction in disease and support the idea that unchecked inflammatory responses promote neurodegeneration. TREM2 is a microglial receptor that stimulates phagocytosis while suppressing inflammation (reviewed in Rohn, 2013). Tanzi suggested that while disease-specific factors might trigger AD or PD, inflammation could exacerbate pathology in TREM2 mutation carriers. “It is likely that across all these neurodegenerative diseases … microglia trigger inflammation,” he said.
Jochen Walter, Harald Neumann, and colleagues at the University of Bonn, Germany, began studying TREM2 in neuroinflammation before it emerged as an AD gene. They noticed that the cell surface receptor shared similarities with other Type-I membrane proteins that are cleaved by the γ-secretase protease complex. In the Journal of Biological Chemistry online, first author Patrick Wunderlich and colleagues reported that TREM2 indeed undergoes a two-step proteolytic cleavage process similar to Aβ precursor protein.
TREM2 consists of an extracellular immunoglobulin-like domain, a transmembrane segment, and a cytoplasmic tail. The researchers found that batimastat, an inhibitor of proteases from both the matrix metalloprotease and A Disintegrin and Metalloprotease (ADAM) families, prevented shedding of the Ig-like domain. The γ-secretase inhibitor DAPT interrupted a second cleavage step. “The data suggest very similar processing as for APP: first ectodomain shedding and then cleavage by γ-secretase,” Walter said. In the presence of DAPT, the Ig-free transmembrane fragments, which would be akin to APP C-terminal fragments, accumulated, as they did in cells expressing a presenilin 1 dominant-negative mutant.
Wunderlich and colleagues further found that DAPT blocked TREM2 signaling through the adapter protein DAP12. The unprocessed transmembrane fragments might act as decoys, binding DAP12 so it cannot interact with full-length TREM2, suggested Carson, who was not involved in the study.
These results imply that while inhibiting γ-secretase might block Aβ production, it could also limit TREM2 signaling. Carson said it is too early to make inferences about Alzheimer’s disease or treatments from these data, but experts who spoke with Alzforum were intrigued to see γ-secretase unexpectedly cleave the product of a second Alzheimer’s gene. “This is exciting, to know that these two pathways are merging,” said Zbigniew Wszolek of the Mayo Clinic in Jacksonville.
Whither Next?
TREM2 has raised many questions for scientists. For starters, Walter asked which ligands interact with it and what cellular changes they induce. TREM2 signaling could affect cytokine release, phagocytosis, or microglial migration, he speculated. In addition, what does the R47H mutation do to TREM2? It might simply render the receptor nonfunctional, or it might alter the ligand binding site so it binds the wrong ligand, suggested Carson.
Guerreiro raised another question: How do mutations in this gene lead to such disparate symptoms as dementia and bone loss? Though hardly the whole answer, the difference has to do with homo- versus heterozygosity. People with two copies of truncated TREM2 genes get the bone disorder as young adults, while carriers of a substitution mutation get Alzheimer’s later in life. Researchers are finding more and more instances where one gene contributes to multiple diseases (for example, see Alzforum News story on Johnson et al., 2010, and Alzforum News story on Tazen et al., 2013).
Researchers are hunting for mutations in DAP12, because, as TREM2’s adaptor protein, it might also harbor variants that boost AD risk. Earlier this year, researchers found that DAP12 regulates molecular networks that are altered in people with AD compared to controls (see Alzforum Live Discussion and Zhang et al., 2013). However, no one has yet found any DAP12 mutations associated with AD. “We have looked, believe me,” said Guerreiro.—Amber Dance.
See also:
TREM2 and Neurodegenerative Disease NEJM 369;16 (1564-70) Reitz C and Mayeux R, for the Alzheimer’s Disease Genetics Consortium, p. 1564-5 Bertram L, Parrado AR, Tanzi RE, the OR is 1.7, p. 1565 Guerreiro R, Hardy J, reply to the issues raised in other letters, p. 1569-70 Jonsson T, Stefansson K, response to other letters, p. 1568-9 Bird T, I saw it first, p. 1568 Rajagopalan P, Hibar DP, Thompson PM, imaging study, p. 1565-7 Benitez BA, Cruchaga C, PD link, p. 1567-8.
References
News Citations
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Mutations in TREM2 Cause Frontotemporal Dementia
- Adding ALS to the Manifestations of VCP Mutations
- One Mutation, Two Diseases in Family with Ataxin-2 Expansion
Webinar Citations
Paper Citations
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):117-27. Epub 2012 Nov 14 PubMed.
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):107-16. Epub 2012 Nov 14 PubMed.
- Ioannidis JP. Why most discovered true associations are inflated. Epidemiology. 2008 Sep;19(5):640-8. PubMed.
- Pottier C, Wallon D, Rousseau S, Rovelet-Lecrux A, Richard AC, Rollin-Sillaire A, Frebourg T, Campion D, Hannequin D. TREM2 R47H variant as a risk factor for early-onset Alzheimer's disease. J Alzheimers Dis. 2013;35(1):45-9. PubMed.
- Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, Cruchaga C. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging. 2013 Jun;34(6):1711.e15-7. Epub 2013 Feb 5 PubMed.
- Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J, Muñoz C, Ramirez-Restrepo M, Gaiteri C, Myers AJ, Caselli RJ, Kosik KS, Reiman EM, Huentelman MJ. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol Aging. 2013 Aug;34(8):2077.e11-8. Epub 2013 Apr 9 PubMed.
- Gonzalez Murcia JD, Schmutz C, Munger C, Perkes A, Gustin A, Peterson M, Ebbert MT, Norton MC, Tschanz JT, Munger RG, Corcoran CD, Kauwe JS. Assessment of TREM2 rs75932628 association with Alzheimer's disease in a population-based sample: the Cache County Study. Neurobiol Aging. 2013 Dec;34(12):2889.e11-3. Epub 2013 Jul 12 PubMed.
- Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, Engelborghs S, Vandenbulcke M, Van Dongen J, Geerts N, Maes G, Mattheijssens M, Peeters K, Cras P, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Cruts M, Sleegers K, BELNEU consortium. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014 Mar;35(3):726.e11-9. Epub 2013 Oct 9 PubMed.
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, . Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012 Jul 6;337(6090):64-9. PubMed.
- Bird TD, Koerker RM, Leaird BJ, Vlcek BW, Thorning DR. Lipomembranous polycystic osteodysplasia (brain, bone, and fat disease): a genetic cause of presenile dementia. Neurology. 1983 Jan;33(1):81-6. PubMed.
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002 Sep;71(3):656-62. Epub 2002 Jun 21 PubMed.
- Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL 3rd, Caselli R, Mackenzie IR, Miller BL, Boczarska-Jedynak M, Opala G, Krygowska-Wajs A, Barcikowska M, Younkin SG, Petersen RC, Ertekin-Taner N, Uitti RJ, Meschia JF, Boylan KB, Boeve BF, Graff-Radford NR, Wszolek ZK, Dickson DW, Rademakers R, Ross OA. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. 2013 Jun 21;8:19. PubMed.
- Lattante S, Le Ber I, Camuzat A, Dayan S, Godard C, Van Bortel I, De Septenville A, Ciura S, Brice A, Kabashi E, . TREM2 mutations are rare in a French cohort of patients with frontotemporal dementia. Neurobiol Aging. 2013 Oct;34(10):2443.e1-2. PubMed.
- Rohn TT. The triggering receptor expressed on myeloid cells 2: "TREM-ming" the inflammatory component associated with Alzheimer's disease. Oxid Med Cell Longev. 2013;2013:860959. PubMed.
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, , Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010 Dec 9;68(5):857-64. PubMed.
- Tazen S, Figueroa K, Kwan JY, Goldman J, Hunt A, Sampson J, Gutmann L, Pulst SM, Mitsumoto H, Kuo SH. Amyotrophic Lateral Sclerosis and Spinocerebellar Ataxia Type 2 in a Family With Full CAG Repeat Expansions of ATXN2. JAMA Neurol. 2013 Aug 19; PubMed.
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 Apr 25;153(3):707-20. PubMed.
Other Citations
Further Reading
Papers
- Guerreiro R, Bilgic B, Guven G, Brás J, Rohrer J, Lohmann E, Hanagasi H, Gurvit H, Emre M. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013 Dec;34(12):2890.e1-5. Epub 2013 Jul 17 PubMed.
- Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, Weale ME, Ryten M. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 Dec;34(12):2699-714. PubMed.
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, , Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013 Jul;16(7):848-50. PubMed.
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013 May 22;78(4):631-43. PubMed.
- Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC, GERAD Consortium, Alzheimer’s Disease Neuroimaging Initiative (ADNI), Alzheimer Disease Genetic Consortium (ADGC), Goate AM. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron. 2013 Apr 24;78(2):256-68. Epub 2013 Apr 4 PubMed.
- Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2010 Jul 12;2(3):e00037. PubMed.
- Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008 Oct;56(13):1438-47. PubMed.
Primary Papers
- Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) by ectodomain shedding and γ-secretase dependent intramembranous cleavage. J Biol Chem. 2013 Nov 15;288(46):33027-36. PubMed.
- Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodríguez-Rodríguez E, López de Munain A, de Pancorbo MM, Pérez-Tur J, Alvarez V, Antonell A, López-Arrieta J, Hernández I, Tárraga L, Boada M, Lleó A, Blesa R, Frank-García A, Sastre I, Razquin C, Ortega-Cubero S, Lorenzo E, Sánchez-Juan P, Combarros O, Moreno F, Gorostidi A, Elcoroaristizabal X, Baquero M, Coto E, Sánchez-Valle R, Clarimón J, dementia genetic Spanish consortium (DEGESCO). Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014 Feb;35(2):444.e1-4. Epub 2013 Sep 13 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Third Rock Ventures
As far as it goes, this paper is quite provocative. The main limitations are that the experiments were done in artificial cell lines and used transfected molecular components. It should be possible, based on these findings, to demonstrate abnormalities of signaling in myeloid cells from individuals (or mice) harboring presenilin 1 mutations. Gamma secretase inhibition should also impair DAP12 signaling in primary myeloid cells. In summary, this study represents an excellent "discovery" exercise that generates important testable hypotheses.
View all comments by Richard RansohoffIcahn School of Medicine at Mount Sinai
I think the most important take-home message from these papers is that each finds evidence that the TREM2 locus is a risk factor for AD and AD-related traits. It would not be surprising for the odds ratio to be slightly lower than reported in the original manuscripts because of the “winner's curse.” However, the meta-analysis of published studies shows the odds ratio still remains above 3, suggesting that carriers of the R47H risk variant have a threefold increase in risk, which is much higher than any of the GWAS hits and similar to carriers of one APOE4 allele. A big difference between the TREM2 variant and APOE4 is the relatively low frequency of the former in the general population; thus, the total number of AD cases attributable to TREM2 will be much lower. The most important aspect of the identification of the TREM2 locus as a risk factor for AD is not the number of cases explained by R47H, or any other variant in TREM2, but the mechanism by which it influences risk for disease. This association highlights the importance of microglia in AD and strongly suggests that lower TREM2 function contributes to AD risk, perhaps through decreased clearance of Aß or regulation of inflammation in the brain.
The paper from Cruchaga et al. hints that R47H may play a role in other neurodegenerative diseases. Papers published in other journals since January have also suggested this possibility, although given the rarity of this and other variants, large meta-analyses will be needed to confirm whether TREM2 variants increase risk for multiple neurodegenerative diseases or whether this risk is specific to AD.
References:
Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC, GERAD Consortium, Alzheimer’s Disease Neuroimaging Initiative (ADNI), Alzheimer Disease Genetic Consortium (ADGC), Goate AM. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron. 2013 Apr 24;78(2):256-68. Epub 2013 Apr 4 PubMed.
�
In general, the frequency of common alleles (i.e., those with frequency greater than 1 to 5 percent) does not differ significantly across populations, but this scenario changes for rare alleles because most of them are population-specific. Furthermore, most of the genetic variants in modern human genomes occur at low frequencies, and consequently the total number of rare alleles vastly outstrips the number of common ones. Importantly, rare variants are more prone to have functional consequences than common variants, and the role of rare variants in disease susceptibility is being established for many traits. However, because rare variants are typically population-specific, population stratification will be an important issue to consider in case-control studies. That is, subtle differences in population structure will result in false-positive and false-negative conclusions.
Next-generation sequencing technologies will allow us to explore the contribution of rare genetic variants to disease risk. However, we are just at the beginning, and I suspect that the way we analyze this data will change somewhat in the near future. First, we will have to make a great effort to use homogeneous populations because, more than ever, subtle differences in population structure will result in false-positive and false-negative conclusions. Second, because rare variants are usually geographically clustered or even limited to specific populations, we will have to build genetic catalogs of rare variants across the globe. Third, we will have to use large sample sizes with accurately classified phenotypes (i.e., using biomarkers, pathologically confirmed diagnosed, and well-defined controls, etc.). Finally, we will have to develop (and use) more sophisticated statistical strategies to evaluate the contribution of rare variants in a particular gene to a common disorder.
TREM2 is a good example of population diversity in the frequency of its risk allele R47H. Even within populations with European ancestry you will see subtle regional differences. For example, the percentage of people carrying the variant are: 0.6 in Iceland; 0.5 in France; 0.54 in Ireland; 0.45 in the United States; 0.15 in several central European countries; 0.1 in Spain. What about Asia and Africa? According to genetic databases such as 1000 Genomes or Exome Variant Server, it is possible that this variant is absent in populations from these regions. This does not mean that this gene is not relevant in these populations, but it could be that other specific, rare variants within TREM2 play a role in non-European populations.
Washington University School of Medicine
In the recent correspondence to the NEJM about the initial TREM2 findings, Bruno Benitez and I reported an association between the TREM2 R47H variant and Parkinson's disease risk. However it looks like neither Dr. Guerreiro nor Dr. Jonsson found the same association in their analysis. An interesting finding reported in these letters is the supplementary table provided by Dr. Hardy, including the R47H minor allele frequency (MAF) for the control populations for all the published reports.
We have since reanalyzed the association of R47H with PD risk using data for that table and including all control datasets in which the R47H variant was directly genotyped or sequenced. In this new analysis our original association remained significant: MAF for R47H in PD cases vs. controls was 0.79 percent vs. 0.38 percent; odds ratio (OR)=2.08, p= 0.03. If we include the data from Rayaprolu et al., in which an additional 1,493 PD samples were genotyped for the R47H variant, then the OR increases to 2.49 with a p=2.31×10-4.
In the reply to our correspondence, Dr. Jonsson indicated that they analyzed 2,730 PD cases and 73,710 controls, and found an OR of 1.24. We supposed that the MAF of the R47H in controls was 0.46 percent based on their original NEJM paper (Jonsson et al., 2013 and performed a meta-analysis including this dataset and the data from our reanalysis as outlined in the paragraph above. In this scenario the R47H variant remained significant, with an OR of 1.32 and p=0.03. Based on the information published by Dr. Jonsson in his reply, it is not clear whether the R47H variant was genotyped or imputed in those 73,710 controls, and what defined controls for this dataset. As they originally published, the MAF of the R47H is quite different in elderly individuals vs. cognitively normal individuals. Therefore, their estimation of the risk associated with the R47H variant in PD may be underestimated.
In summary, based on these analyses we feel confident about the association of the TREM2 R47H variant with PD risk. However, if both Drs. Guerreiro and Jonsson would publish or share their data, a more rigorous meta-analysis could be performed.
References:
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL 3rd, Caselli R, Mackenzie IR, Miller BL, Boczarska-Jedynak M, Opala G, Krygowska-Wajs A, Barcikowska M, Younkin SG, Petersen RC, Ertekin-Taner N, Uitti RJ, Meschia JF, Boylan KB, Boeve BF, Graff-Radford NR, Wszolek ZK, Dickson DW, Rademakers R, Ross OA. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. 2013 Jun 21;8:19. PubMed.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):107-16. Epub 2012 Nov 14 PubMed.
Make a Comment
To make a comment you must login or register.