Innate Immune Cells Enlisted to Clear Amyloid, Fight Disease
Quick Links
This concludes a two-part story. Read part one here.
As researchers flesh out connections between the brain and the immune system, they are beginning to understand the role microglia play in disease. Prior research had blamed overzealous microglia for spewing inflammatory molecules that damage brain tissue, but recent studies suggest that AD may progress more quickly when these brain-resident phagocytes become sluggish and less responsive as they age (see part one of the series). Both scenarios may be true. At the Society for Neuroscience’s annual meeting, held November 9–14 in San Diego, several presentations focused on strategies to reinvigorate the brain’s innate immune cells. Researchers also presented approaches that harness innate immune pathways to reduce brain Aβ while toning down potentially harmful inflammatory responses. The new data raise hopes of capitalizing on beneficial glial cell responses, but don’t yet fully answer the question of exactly what those are.
The idea that innate immune activation might help in AD found early support in reports that blood-derived macrophages cleared brain Aβ in mouse models of AD. In 2008, researchers led by Terrence Town, now at the University of Southern California, Los Angeles, reduced AD-like pathology in Tg2576 mice by crossing them to a transgenic strain whose macrophages lack anti-inflammatory TGFβ signaling (Jun 2008 news story). At SfN, Tara Weitz, a postdoc in Town’s lab, reported on a pharmacological version of the TGFβ-blocking strategy.
Weitz showed that the commercially available TGFβ inhibitor SB-505124 prompted primary mouse macrophages to chew up Aβ in cell culture. When she treated 14-month-old Tg2576 mice with a four-month regimen of biweekly intraperitoneal injections, the inhibitor slowed further amyloidosis in these already plaque-burdened animals. Moreover, activated, i.e., CD45-positive, peripheral macrophages infiltrated blood vessels in the cingulate cortex, the hippocampus, and the entorhinal cortex—regions of the brain where Aβ accumulates in people.
However, when Weitz and colleagues administered the inhibitor to 16-month-old transgenic AD rats, which also have a high amyloid burden (Cohen et al., 2013), plaque load hardly budged. Though disappointing, the results “confirmed our hypothesis that the rat may be a more stringent AD model that is harder to treat,” Weitz told attendees (see Aug 2010 conference story). Many treatment strategies that appear to cure mice of AD pathology have failed in human clinical trials (e.g., Aug 2010 news story; Dec 2009 news story).
The researchers are getting more promising results loading the inhibitor into nanoparticles developed by Tarek Fahmy, a medicinal chemist at Yale University, New Haven, Connecticut. In addition to carrying the TGFβ inhibitor, these 100- to 200-nm-wide particles carry the green dye Coumarin-6 for easy tracking. In cell cultures, the nanoparticles readily entered wild-type mouse macrophages, lingering in the cells about a week. “The macrophages ‘see’ the nanoparticles as trash, and voraciously gobble them up,” Town said. “It’s an elegant physiologic system requiring no surface labeling or targeting.”
The researchers tested the SB-505124-laden nanoparticles in 8-month-old AD rats, which had moderate amyloid load. When the particles were given weekly as a footpad injection, for six months, peripheral macrophages took them up. The macrophages then homed in on amyloid-laden cortical and hippocampal areas, the researchers found. Compared with control AD rats treated with drug-free nanoparticles, treated animals had fewer plaques and more activated macrophages as judged by CD11b expression.
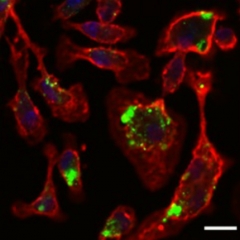
Feasting Glia
Peripheral macrophages (red) devour nanoparticles (green) loaded with TGF inhibitor. Image courtesy of Tara Weitz.
At this point, it is unclear if the nanoparticles are more effective than the naked inhibitor, or whether starting injections at a much younger age made a difference, Weitz said. In addition to looking at behavioral readouts in the treated cohort, her team plans to administer SB-505124 nanoparticles to younger rats in a prevention paradigm.
The Town lab also tests other strategies to activate microglia. The researchers have bred PSAPP double-transgenic mice onto an interleukin (IL)-10-deficient background. Because IL-10 quells inflammation, “We’re essentially releasing brakes on innate immunity,” Town said. As reported earlier this year at a conference in Bonn, Germany, the IL-10-negative animals activate more microglia and accumulate less amyloid than PSAPP mice (see Mar 2013 conference story). At SfN, Marie-Victoire Guillot-Sestier of the Town lab showed that the IL-10-deficient PSAPP mice outperform controls in cognitive tests, steering clear of open fields and more readily recognizing novel objects. Without IL-10, the mice maintain better synapses, as measured by synaptophysin levels. The Town lab has a similar IRAK-M (IL-1 receptor-associated kinase M)-deficient PSAPP strain; synapse and cognitive data on that is forthcoming, Town said.
Paramita Chakrabarty and Todd Golde of the University of Florida, Gainesville, have data that complement Town’s work. Rather than blocking IL-10, these researchers used adeno-associated viral vectors to overexpress this cytokine in the brains of two AD mouse strains. TgCRND8 mice received the transgene injection at birth and Tg2576 mice at 8 months of age, when amyloidosis is well underway. Relative to vehicle controls, IL-10-overexpressing TgCRND8 mice racked up more plaques, performed poorly on fear-conditioning tests, and their microglia appeared less phagocytic, Golde said. The IL-10-overexpressing Tg2576 mice also had higher plaque burden compared to regular Tg2576.
Furthermore, Chakrabarty presented a strategy that clears brain Aß in TgCRND8 mice while dampening microglial activity. The researchers enlisted toll-like receptors (TLRs); these are surface receptors on immune cells that recognize proteins typically found on microbes. TLRs also bind Aß, and microglia from TLR4 knockout mice cannot phagocytose the peptide (Sep 2009 news story on Reed-Geaghan et al., 2009; see Boutajangout and Wisniewski, 2013 review).
To curb cerebral amyloidosis, Chakrabarty and colleagues engineered soluble forms of TLRs 2, 4, 5, and 6 that contain the receptors' extracellular Aß-binding region but lack their cytoplasmic signaling domains. The truncated molecules behave as decoys, neutralizing Aß without stimulating unwanted inflammatory responses. Widely used anti-inflammatory drugs function that way, for example the tumor necrosis factor decoy receptor etanercept. TgCRND8 mice that had received injections of truncated soluble TLR4 (sTLR4) or soluble TLR5 (sTLR5) at birth grew up accumulating fewer plaques and less soluble Aß in the brain. In addition, fewer glial cells surrounded plaques in the brains of sTLR4- and sTLR5-expressing mice, and those that did had blunted responses as judged by expression of the glial activation marker GFAP.
In culture, the decoy receptors blocked the toxic effects of aggregated Aß42 and α-synuclein, and ELISAs confirmed they did so by binding the protein aggregates, Chakrabarty reported. In future experiments, the researchers hope to determine if the soluble TLRs can reach the brain if given peripherally and prevent cognitive deficits. In addition, Golde’s group is testing whether the approach could help AD mice with amyloidosis already underway, and if it can alleviate pathology and behavioral deficits in stroke models and transgenic mice overexpressing tau or α-synuclein.
Active, Idle, or Simply Different?
While the sTLR approach seems to suggest otherwise, broadly speaking “there is an emerging concept that microglia in amyloidosis mouse models are less active than they should be,” David Morgan of the University of South Florida, Tampa, told Alzforum. Their suppressed state “is somehow induced by amyloid deposition, and relieving the suppression enables circulating monocytes to go into the brain and clear out the amyloid,” Morgan said. However, he cautioned that activating microglia may not be a viable treatment for AD as it worsens tau pathology in some mouse studies (Ghosh et al., 2013; Oct 2010 news story; see also Lee et al., 2013 review).
Furthermore, it is still hard to figure out which immune cells to activate, and how. Researchers had looked to distinguish harmful and helpful microglial subtypes, dubbed M1 and M2, based on their expression of pro- and anti-inflammatory cytokines, but now some scientists consider these designations inadequate (see Mar 2013 conference story). “I think we need to drill down further to identify the specific protein markers that go up under conditions where we’re clearing amyloid from the brain,” Morgan said.
As outlined on an SfN poster by Kevin Doty, who studies bioinformatics in the Town lab, scientists are starting to make inroads in this direction. Hypothesizing that targeting specific immune mediators is better than generally dialing inflammation up or down, Doty tried to establish why PSAPP/IL-10-negative mice accumulate less Aß. He profiled transcription in whole-brain tissue with RNA sequencing and network analysis. He found that the mice downregulated a handful of genes involved in innate immune regulation. That list includes the microglial receptor TREM2, which was recently identified as an AD risk gene (Nov 2012 news story). “While the link between the rare TREM2 variant and AD risk remains poorly understood, one possible explanation for Doty’s findings is that TREM2 inhibits Aß phagocytosis by microglia,” said Town. The work also suggests that IL-10 is involved in TREM2 regulation “The goal is to identify a ‘fingerprint’ for beneficial innate immune activation to aim for with Alzheimer’s immunotherapies,” Town told Alzforum.
Applying a similar strategy on purified microglia from wild-type mice, Joseph El Khoury of Massachusetts General Hospital, Charlestown, and colleagues identified 100 genes that are differentially expressed in aging microglia. TREM2 made that list, too—as did another AD risk gene, CD33, and DAP12, an adaptor protein that mediates TREM2 signaling but has no known genetic connection to AD yet. El Khoury reported some of this data at the recent Venusberg meeting (see Mar 2013 conference story), and in full in an October 27 Nature Neuroscience paper (Hickman et al., 2013). “These studies are telling us there are many different types of activated microglia,” Town said. While El Khoury is finding that microglia change their activation profile with age, “we are finding that they change in the context of AD pathology.”
Other scientists at SfN presented research aimed at identifying microglial-specific markers. Mariko Bennett of Ben Barres’ lab at Stanford School of Medicine, Palo Alto, California, isolated RNA out of microglial-enriched, CD45-positive pools of cells from healthy mice. They identified transmembrane protein 119 (TMEM119) as a novel microglia-specific marker. About 93 percent of myeloid cells in the brain and spinal cord express TMEM119 RNA, and the protein is highly restricted to microglia.—Esther Landhuis
References
News Citations
- Glial Cells Refine Neural Circuits
- Macrophages Storm Blood-brain Barrier, Clear Plaques—or Do They?
- Honolulu: Monkey, Rat Join Menagerie of Mammalian AD Models
- Lilly Halts IDENTITY Trials as Patients Worsen on Secretase Inhibitor
- Paper Alert—Phase 3 Tarenflurbil Data Published
- Blessing or Curse? Peripheral Cytokines in the Brain
- New Pathways With Promise in AD—An Inflammatory Statement?
- Paper Alert: Fractalkine Receptor Hits Aβ, Tau, in Opposite Ways
- Microglia Activation—Venusberg Meeting Questions M1, M2 Designations
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
Paper Citations
- Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013 Apr 10;33(15):6245-56. PubMed.
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009 Sep 23;29(38):11982-92. PubMed.
- Boutajangout A, Wisniewski T. The innate immune system in Alzheimer's disease. Int J Cell Biol. 2013;2013:576383. Epub 2013 Oct 2 PubMed.
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, Laferla FM, Olschowka JA, O'Banion MK. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci. 2013 Mar 13;33(11):5053-64. PubMed.
- Lee DC, Rizer J, Hunt JB, Selenica ML, Gordon MN, Morgan D. Review: Experimental manipulations of microglia in mouse models of Alzheimer's pathology: activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol. 2013 Feb;39(1):69-85. PubMed.
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013 Dec;16(12):1896-905. Epub 2013 Oct 27 PubMed.
Other Citations
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.