Mitochondrial Jetsam Spurs Neurodegeneration
Quick Links
Mitochondrial fragments, jettisoned by stressed cells, create trouble for other cells, according to a study in the September 23 Nature Neuroscience. Researchers led by Daria Mochly-Rosen at Stanford University School of Medicine report that when microglia spat out damaged mitochondria, these cast-offs inflamed astrocytes, which in turn expelled their own mitochondrial fragments. Jetsam from either cell sickened neurons as well, limiting their energy production. Conversely, an inhibitor of mitochondrial fission protected astrocytes and neurons from the effect of externally added mitochondrial fragments, suggesting that mitochondrial fragmentation cascades from cell to cell. Inhibiting fission prevented gliosis and protected neurons in the 5xFAD model of Alzheimer’s pathology as well. Curiously, adding whole, functional mitochondria to neuronal cultures also mitigated the damage from fragmented organelles. “We need to pay attention to mitochondria not only as powerhouses, but also as signaling organelles that communicate between cells,” Mochly-Rosen proposed.
- Stressed microglia and astrocytes spit out mitochondrial fragments, which harm cells.
- Inhibiting mitochondrial fission protects neurons.
- Extracellular whole, functional mitochondria somehow protect neurons.
Derek Narendra at the National Institute of Neurological Disorders and Stroke, Bethesda, Maryland, noted that mitochondria are ancient bacterial invaders of eukaryotic cells, but are tolerated by the body because they are sequestered inside cells. Once released, their proteins and other macromolecules may trigger inflammation. “Mitochondria have long been recognized to be important players in neurodegeneration, but the precise vulnerability has been unclear. This and other recent work suggest that the key may be recognition of these ancient mitochondrial invaders by the immune system,” he wrote to Alzforum (full comment below).
Mitochondrial damage accumulates during aging and Alzheimer’s disease (May 2001 news; Jul 2009 news; Aug 2013 news). Faulty mitochondria mark other neurodegenerative disorders as well, including amyotrophic lateral sclerosis, Parkinson’s, and Huntington’s (Reddy, 2014; Jul 2015 news; Jun 2016 news). Mochly-Rosen and colleagues previously characterized a seven-amino-acid peptide, P110, that prevents aberrant, stress-induced mitochondrial fission while sparing physiological organelle division (Qi et al., 2012). In mouse models of Huntington’s and ALS, P110 improved survival and motor skills, while in 5xFAD mice it improved memory, suggesting broad neuronal protection (Guo et al., 2013; Joshi et al., 2018; Joshi et al., 2018).
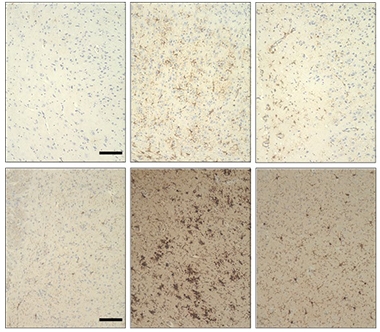
Dousing Inflammation. 5xFAD mice develop more astrogliosis (top middle) and microgliosis (bottom middle) than age-matched wild-type (left). An inhibitor of mitochondrial fission (right) protects. [Courtesy of Joshi et al., Nature Neuroscience.]
Exactly how P110 protected neurons in these mouse models was unclear. To find out, first author Amit Joshi analyzed the brains of treated mice. He found a dramatic suppression of microglial and astrocyte activation in all three models (see image at right). To find out if this was a direct effect of P110, Joshi added the inhibitor to microglia expressing a 73-amino-acid polyglutamine expansion (Q73) that causes mitochondria to malfunction and fragment. P110 treatment reduced mitochondrial fission, boosted ATP production, and lowered reactive oxygen species. It also halved production of proinflammatory cytokines, including tumor necrosis factor-α and interleukin-1β. P110 similarly benefitted microglia expressing superoxide dismutase carrying a familial ALS mutation or treated with oligomeric Aβ42.
How might microglia in these models affect other cell types? The authors added media from Q73 microglia to mouse primary astrocyte cultures. In response, the astrocytes pumped out TNF-α and IL-1β. Their mitochondria became dysfunctional and fragmented, and 75 percent more astrocytes died. However, if the Q73 microglia were treated with P110 a day before their media was collected, these effects were largely neutralized and more of the astrocytes survived. Adding P110 directly to astrocytes also protected them from the Q73 microglia-conditioned media.
Likewise, conditioned media from microglia carrying Q73 or SOD1 mutations suppressed cellular respiration and cut short survival of primary neurons. So did media from astrocytes that had been treated with the microglia-conditioned media. Once again, treating either glial culture with P110 first lessened the neuronal damage. P110 added directly to neurons also protected them, suggesting that fission of their mitochondria drove the effects on their health. Taken together, the data implied that fragmentation of mitochondria causes microglia and astrocytes to release factors that can somehow damage mitochondria in other cell types.
The authors wondered if those released “factors” might be mitochondria themselves. Scientists have documented release of intact mitochondria into the extracellular space, and even their uptake by other cells (Falchi et al., 2013; Hayakawa et al., 2016).
Confirming this, the authors found intact functional mitochondria in media from healthy microglial cultures. Media from Q73 microglia cultures contained the same total number of mitochondria as media from the healthy cultures, but only half as many were whole and functional. At the same time, the amount of free-floating mitochondrial proteins in Q73 culture medium rose, suggesting the organelles were leaking contents. Treating Q73 microglial cultures with P110 bumped the number of functional mitochondria almost back to that of control cultures. Microglia carrying ALS mutations or treated with lipopolysaccharide also released damaged mitochondria into the cell medium, and again added P110 ameliorated this. These damaged extracellular mitochondria were a major source of toxicity to other cells, the authors found. Filtering out the organelles protected neurons from conditioned medium to the same extent as did pretreatment of the glial cultures with P110.
Notably, the functional mitochondria in cell media helped neurons. When the authors removed whole mitochondria from control microglial media and then added it to neurons, more cells died than when complete medium was added.
“We were surprised that extracellular mitochondria can affect neighboring cells,” Mochly-Rosen told Alzforum. While the damaged organelles may trigger inflammation by activating microglia or astroglia, their direct effect on neurons remains puzzling, as does the effect of whole mitochondria. Mochly-Rosen is investigating the idea that neurons take up whole or fragmented organelles, with the former bolstering cellular respiration and the latter spreading damage. She is also studying how mitochondria are released from cells in the first place, and exploring a potential role for extracellular mitochondria in other disorders, such as sepsis and cardiac arrest. “It’s possible we’ve found a common mechanism by which cells communicate under pathological stress versus under normal conditions,” she said.
Could reducing aberrant mitochondrial fission be a therapeutic strategy for such conditions? Mochly-Rosen believes so. She noted that P110 enters the brain, shows up in neurons, and appeared safe in a five-month mouse toxicity study. Additional safety studies are ongoing. However, the peptide degrades rapidly, suggesting a more stable molecule might have stronger effects and work better for people. Other work supports the idea that cleaning up damaged mitochondria could help treat Alzheimer’s pathology (Feb 2019 news).—Madolyn Bowman Rogers
References
Research Models Citations
News Citations
- Mitochondrial Damage in Alzheimer's Disease
- Mitochondrial Break-up: Alzheimer’s Alters Fusion, Fission
- Studies Suggest Mitochondria Changes Precede Aging, Alzheimer’s
- Juvenile Lysosomes and Worn-Out Mitochondria Clog Axons in ALS Model
- A Sinister Side to α-Synuclein—Blocking Mitochondrial Protein Import
- Could Disposing of Damaged Mitochondria Treat Alzheimer’s Disease?
Paper Citations
- Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington's disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today. 2014 Jul;19(7):951-5. Epub 2014 Mar 28 PubMed.
- Qi X, Qvit N, Su YC, Mochly-Rosen D. Novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2012 Dec 13; PubMed.
- Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington's disease-associated neurodegeneration. J Clin Invest. 2013 Dec 2;123(12):5371-88. Epub 2013 Nov 15 PubMed.
- Joshi AU, Saw NL, Vogel H, Cunnigham AD, Shamloo M, Mochly-Rosen D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol Med. 2018 Mar;10(3) PubMed.
- Joshi AU, Saw NL, Shamloo M, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer's disease. Oncotarget. 2018 Jan 19;9(5):6128-6143. Epub 2017 Dec 22 PubMed.
- Falchi AM, Sogos V, Saba F, Piras M, Congiu T, Piludu M. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem Cell Biol. 2013 Feb;139(2):221-31. Epub 2012 Oct 30 PubMed.
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016 Jul 28;535(7613):551-5. PubMed.
Further Reading
News
- Protein Screen Links Mitochondrial Regulator to Alzheimer’s Disease
- New Triple Transgenic Shows Mitochondrial Damage by Tau, Aβ
- Evidence Mounts That Mitochondrial Gene Is Bona Fide ALS, FTD Risk Factor
- So Immature—Aβ Stymies Mitochondrial Protein Processing
- Mislocalized Mitochondria Sufficient for Motor Neuron Malaise
- Energy Crisis: ATP Deficiency Dooms Motor Neurons in Computer Model
- Mitochondrial Mutation Linked to Syndrome With ALS-FTD Features
- Gene Links Childhood Neurodegeneration to Mitophagy
- Protein Destroying Muscle, Bone, Nerves Parks on Mitochondria
Primary Papers
- Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, Mochly-Rosen D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 2019 Oct;22(10):1635-1648. Epub 2019 Sep 23 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
National Institutes of Health
This very interesting article from lead author Amit Joshi adds to growing evidence that damaged mitochondria may propagate neurodegeneration by triggering neuroinflammation. Mitochondria are bacterial in origin. Their unique proteins, lipids, and circular DNA are usually protected from the innate immune system by two membranes within the cell. When damaged they may release bacterial-like macromolecules from this privileged space to be recognized by the immune system and incite inflammation. In the context of Parkinson’s disease, two recent papers in Nature (Sliter et al., 2018; Matheoud et al., 2019) demonstrated that damaged mitochondria in mice lacking the PD gene PINK1 trigger neuroinflammation through recognition of released mitochondrial DNA and display of mitochondrial antigens.
This article from Joshi et al. expands on this theme, showing that in microglia, the brain’s resident immune cells, mitochondria are damaged by neurotoxic proteins such as Aβ and that the damaged mitochondria release fragments into the extracellular milieu, activating astrocytes and causing neuronal death. Mitochondria have long been recognized to be important players in neurodegeneration, but the precise mitochondrial vulnerability has been unclear. This and other recent work suggest that the key may be recognition of these ancient mitochondrial invaders by the immune system.
References:
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018 Sep;561(7722):258-262. Epub 2018 Aug 22 PubMed.
Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, Ducrot C, Laplante A, Bourque MJ, Zhu L, Cayrol R, Le Campion A, McBride HM, Gruenheid S, Trudeau LE, Desjardins M. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1-/- mice. Nature. 2019 Jul;571(7766):565-569. Epub 2019 Jul 17 PubMed.
National Institute on Aging
The work from Joshi et al. provides an interesting mechanistic hypothesis to explain how multiple cell types may contribute to several different neurodegenerative diseases. That non-neuronal cells contribute to neuronal cell loss probably has been best established in SOD1-ALS but is also likely to be true in other diseases. Given that the genes influencing monogenic forms of AD, ALS, and HD are all different and are associated with different phenotypes in humans and the mouse models used here, my interpretation of the data presented here is that the mitochondrial pathway is likely to be a late common pathway for neuronal damage rather than an initiating event.
It might therefore be interesting to look at the role of mitochondrial-mediated activation in those diseases that appear to spread throughout the brain. It would also be important to establish whether cell-type restricted expression of P110 in vivo would be sufficient to block cell death as it does in the primary culture experiments reported here.
Make a Comment
To make a comment you must login or register.