Energy Crisis: ATP Deficiency Dooms Motor Neurons in Computer Model
Quick Links
For the first time, ALS researchers have used computational modeling to probe why motor neurons are so susceptible to degeneration. They report in the July 31 Neuron online that even a slight dearth of ATP pitches the in silico neurons into a deadly feedback loop, in which sodium-potassium pumps demand more and more energy from an ever-dwindling cellular supply. In a real neuron, this presumably would be fatal, said study co-author Serge Przedborski of Columbia University in New York.
To the authors’ knowledge, no other researchers have attempted to model neurodegenerative disease in a similar cell-based computational system. First author Gwendal Le Masson visited Columbia University during a sabbatical from the University of Bordeaux, France. While in New York, he developed the model in collaboration with Przedborski, who provided guidance on the neuroscience, and Larry Abbott, who furnished the computational expertise. The authors modified a generic neuron, in a commonly used simulator called NEURON (Hines and Carnevale, 1997), so that it would resemble a motor neuron. They lengthened its axon and tweaked the distribution of ion pumps for sodium, potassium, and calcium to match the known parameters of real motor neurons.
Przedborski is interested in the contribution of mitochondria to ALS, hence the researchers decided to test the hypothesis that a crimped supply of ATP, such as might result from sick mitochondria, could affect a neuron’s electrophysiological properties. While this would be a complicated question in a living system, in the computer Le Masson was able to focus on just the ATP and ion pumps.
Taking away just 15 percent of the computer neuron’s ATP proved disastrous, affecting both sodium-potassium transporters and calcium homeostasis. The ATP-dependent sodium potassium pumps normally swing into action right after a nervous impulse passes, restoring the cell membrane to its proper polarity. With less ATP available to them, the transporters quickly became overwhelmed. The neuron was left less than fully repolarized, making it more prone to fire and depolarize again. This led to what the authors termed a “deadly loop” as the ion pumps kept trying to repolarize the membrane, sucking up more ATP, leading to even lower ATP stores and worsening the situation.
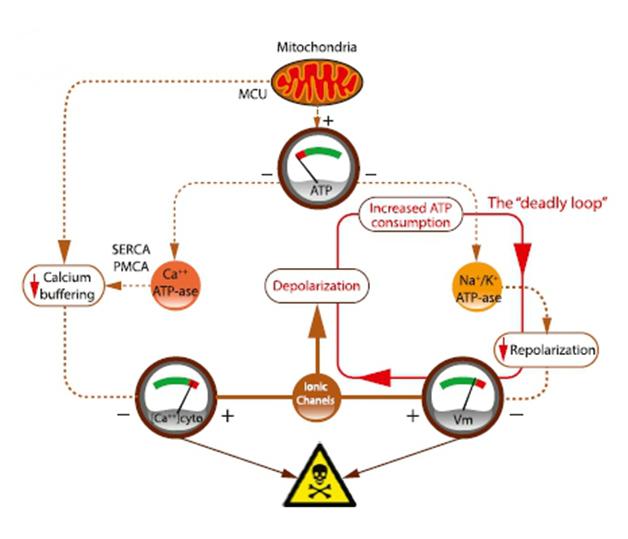
When ATP stores run low, ion pumps cannot keep up with the neuron’s needs. Dashed lines indicate processes impeded by diminished ATP. Sodium-potassium pumps cannot repolarize the cell membrane after an action potential, trapping the neuron in a “deadly loop.” [Image courtesy of Neuron, Le Masson et al., Figure 8B.]
Calcium transporters also need ATP to pump the ion out of the cytosol, into the ER, or outside the cell. When Le Masson dropped the ATP, he observed that calcium buffering faltered. This could have numerous negative consequences for the cell, Przedborski said, as a variety of enzymes and regulatory factors are sensitive to calcium concentrations. Przedborski hypothesized that, over a lifetime, lowered ATP levels could sensitize a person’s motor neurons to degeneration.
Even among motor neurons, some are more vulnerable than others. The energetics of the computer neuron explain why this might be. Le Masson modeled so-called fast-fatigable motor neurons, which are particularly prone to degeneration in ALS, and slow motor neurons, which survive longer. The fast-fatigable cells are larger and more branched. They consume more ATP per action potential than the slow ones and, sure enough, they were particularly susceptible to the deadly loop.
Giovanni Manfredi of Weill Cornell Medical College in New York commented that he found the work convincing, although these potential mechanisms for motor neuron degeneration are not new. What Manfredi found new was the author’s next simulation of how motor neurons might “die back” from axon terminals, as is observed in animal models. Even when the ATP supply was down only at axon terminals, the effects eventually reached the cell body and doomed the whole cell.
“This model can merge many observations relative to the pathogenesis of ALS,” Manfredi said. “I hope these types of ideas … rekindle a bit of interest in global energy problems.” While researchers suspect that mitochondrial defects and altered bioenergetics contribute to ALS (see Jan 2012 news story; Dec 2010 news story), this study is the first to demonstrate how a cellular energy crisis could specifically damage neurons, Przedborski said. Of course, any computational study carries the caveat of being artificial, he added. Though Abbott carefully checked the program to make sure that the computer neuron “behaved” like a real one, Przedborski said, “It is absolutely still possible that we have omitted some glaring elements here, and what we have modeled will never happen.” To put their results to the test, Przedborski and Le Masson are now performing electrophysiology and calcium imaging on cultured neurons.
Przedborski hopes to examine other questions in the computer, as well. “Everything that you can conceptualize in a mathematical manner can be [modeled],” he said. He hopes to whet other researchers’ appetites to try computational modeling of neurodegenerative disease themselves. “This was a lot of fun, and really powerful to test all sorts of crazy ideas in a simplified system,” he said.—Amber Dance
References
News Citations
- More Mitochondrial Mayhem in ALS Motor Neurons, Muscles?
- San Diego: Mutant SOD1 Bumps Mitochondrial Current Up—Or Down?
Paper Citations
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997 Aug 15;9(6):1179-209. PubMed.
External Citations
Further Reading
Papers
- Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput Neurosci. 2013;7:13. PubMed.
- Jalil S, Grigull J, Skinner FK. Novel bursting patterns emerging from model inhibitory networks with synaptic depression. J Comput Neurosci. 2004 Jul-Aug;17(1):31-45. PubMed.
- Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2004 Nov-Dec;19(6):345-52. PubMed.
- Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001 Spring;106(1):27-36. PubMed.
- Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012 Sep;342(3):619-30. PubMed.
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002 Aug 16;277(33):29626-33. PubMed.
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012 Jun 3; PubMed.
News
- NIH Funds Prevention Trials and Translational Studies
- An Ultra-High-Resolution, 3D View of the Human Brain
- Mitochondria Stumble Their Way Along Axons in ALS Model
- Mutant Meddling in Mitochondria Partly Mimics ALS Pathology
- ALS: Mutant Enzyme Does Not Hang Around, But Does Find Mitochondria
- New Theory for Some ALS Cases—SOD1 Plugs Cell Power Plants
Primary Papers
- Le Masson G, Przedborski S, Abbott LF. A computational model of motor neuron degeneration. Neuron. 2014 Aug 20;83(4):975-88. Epub 2014 Jul 31 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.