Genetics Propels DIAN Toward Therapies
Quick Links
Each year, the DIAD family conference held in advance of the Alzheimer's Association International Conference features a science update, and this year the focus was on genetics. DIAN families know that the modern era of Alzheimer’s research started when blood given by their parents enabled the discovery of mutations in APP, presenilin 1 and 2 (Goate et al., 1991; Sherrington et al., 1995; Rogaev et al., 1995; Levy-Lahad et al., 1995). They may not know that Alzheimer’s genetics research today continues to draw on dominantly inherited AD. “The discoveries go back to the samples you donated in the 1980s, but they really do not end there,” Alison Goate told the families. Goate, who was at University College London when she discovered the first APP mutation, is now at Icahn School of Medicine, Mount Sinai, New York.
- Twenty-eight years after first gene was found, dominantly inherited AD still devastates families.
- Genes discovered for late-onset AD influence age of symptom onset in APP, PSEN mutation carriers.
- Geneticists are on the hunt for protective genes that delay onset.
By the late 1990s, after scientists had fingered ApoE as a risk gene for late-onset AD (Corder et al., 1993), progress in AD genetics overall slowed to a crawl for a decade or so. Scientists knew that APP and presenilin mutations caused rare, dominantly inherited AD on the one hand, and that ApoE4 explained a small piece of the common, late-onset form of AD on the other hand. However, in between these two was a large unexplained gap they were unable to close with the techniques at the time. They were stuck. Then, with the advent of GWAS and new sequencing and analysis methods, AD genetics research revived—and once again, dominantly inherited mutations are part of the progress.
Nowadays, scientists use the term “genetic architecture” to convey that the genetic basis of Alzheimer’s disease is more like a complex assembly of many elements than the product of a single gene. The respective “power” of different gene mutations, or variants, falls onto a spectrum. On one end are dominantly inherited variants in a single gene that are powerful enough to cause AD in early adulthood; they are rare. On the other end are variants that raise a person’s Alzheimer’s risk by only a smidgen; they occur commonly among the different world populations. In between these extremes of severity and frequency, however, are known to be many variants in many additional genes that are more or less pathogenic and more or less frequent. This in-between group contains ApoE and many other genes, including Trem2, CD33, and Sorl1, for example (Campion et al., 2019). For every given person, it is their inherited unique combination of variants from this collection of genes that determine if and when they develop dementia.

This much-cited image from a paper by Teri Manolio and colleagues depicts how disease variants of genes fall onto a spectrum defined by strength (effect size) and frequency in the population. DIAD mutations would crowd the top left corner. Many of the weaker, and more common, gene variants that are still being discovered likely influence when Alzheimer’s dementia develops in a given APP and PSEN mutation carrier. [Manolio et al., Nature, 2010.]
How does this play out in reality? For example, the 501 families who are in DIAN all have a known pathogenic mutation in APP, PSEN1, or PSEN2, and almost all carriers get the disease. Even so, age of onset varies from one person to the next. This is especially true of PSEN1, which, at 392 families with 177 different mutations, is the most commonly mutated gene in DIAD. While many mutations cause symptoms by one’s 40s, the spread across PSEN1 mutations ranges from age 20 to 80, and even within a given family’s single mutation, onset can differ by a decade. Indeed, when scientists looked among non-DIAN families who had clusters of late-onset AD (LOAD), 3.4 percent of these people turned out to carry a “DIAD” mutation (e.g., Cruchaga et al. 2012). This means that other genes influence when a pathogenic APP or presenilin mutation breaks through in a person, Goate said.

The age when symptoms start ranges widely for autosomal-dominant mutations. [Courtesy of Ryman et al., 2014. © American Academy of Neurology]
One such gene is ApoE, insofar as APP/PSEN mutation carriers with the ApoE2 variant tend to get the family disease later than relatives with the ApoE4 variant. Another AD risk gene that preoccupies scientists is TREM2. It encodes a receptor protein sitting on microglia, the immune cells of the brain. What TREM2 risk variants do in DIAN families is unknown, but it is known that they speed up amyloid plaque formation in the brains of mice made to express DIAD mutations. Bits of the TREM2 protein are detectable in CSF of DIAN participants, and levels rise as people approach the symptomatic stage. “TREM2 probably influences progression of your family disease. The wide spread in age at onset that we see may be partly due to which TREM2 variant a person has,” Goate said.
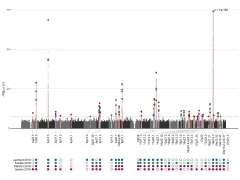
Colored dots on this Manhattan plot represent spots in the genome where geneticists have identified Alzheimer’s risk. To date, they know of 41 such loci. [Courtesy of Shea Andrews & Brian Fulton-Howard.]
Besides ApoE and TREM2, many additional AD risk genes may play into age at onset. In toto, geneticists to date have identified 41 spots, or loci, along the human genome that harbor risk variants for late-onset AD (see image above). When they compute all of these variants into one number—called a polygenic risk score, or PRS—they can see that this score partially accounts for the age at onset of dominantly inherited AD. In other words, the PRS score does not determine whether a DIAN participant will develop AD. That is largely due to the powerful APP or presenilin mutations. Rather, the other AD genes combined affect at what age that happens (Cruchaga et al., 2018).
Using blood from both DIAN and API participants, geneticists are hot on the trail of protective genes. Addressing DIAN families in L.A., Ken Kosik of the University of California, Santa Barbara, emphasized that no gene mutation is 100 percent deterministic. Scientists know that besides ApoE2, certain variants of the genes TMEM106B, phospholipase C, Rab10, and the Icelandic APP mutation delay Alzheimer’s onset (May 2019 news; Tavana et al., 2019; April 2016 news). And they are sure that more are yet to be found.
The DIAN families include several “escapees” who remain cognitively healthy long after their family’s age at onset. One, Doug Whitney, is a regular at the family meeting; this year again, he was as well as ever despite his presenilin 2 “Volga German" mutation. The Colombian cohort includes at present seven carriers of the E280 mutation whose dementia did not start until old age, even though it usually begins in the 40s. Kosik and others are searching for the genes that held their disease at bay. One such gene was grist for the rumor mill at AAIC; its publication is currently embargoed but due out soon. About the others, all Kosik knows so far is that their protective variants do not lie in protein-coding regions. “They are more complicated to find, but we will,” Kosik said. Noncoding mutations tend to work by controlling the expression of other genes.
To understand gene expression in AD, DIAN is proving important in genomics research, as well. Genomics takes the study of genetics beyond a single gene. For example, scientists compare brain tissue from dominantly inherited to that of late-onset AD, and ask in which particular cell types a disease mutation does its damage. Is it neurons? Is it glial cells? At the DIAD family meeting, Goate cited studies by WashU’s Carlos Cruchaga, Oskar Harari, and others. They use pieces of postmortem brains donated by participants in DIAN and other AD studies, physically separate out the nuclei of the different cell types, and quantify which genes each individual cell had been expressing, and in what amounts, while the person was still alive. This is a step forward from mushing up bits of brain tissue and analyzing all the component cell types together, an older practice that obscured big differences between them.
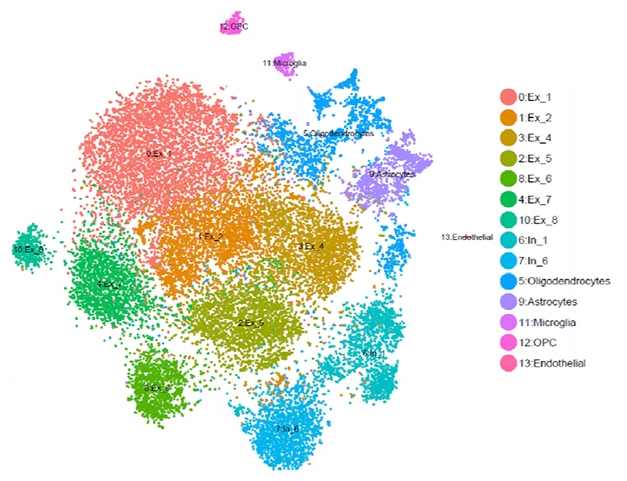
Fewer Neurons in ADAD? An example of single-cell RNA analysis comparing brain tissues from a PS1 mutation carrier and noncarriers. Software named after the French pointillist painter Georges Seurat sorts gene-expression data into clusters. In this study, the carrier had fewer excitatory neurons (red, ochre, green) than noncarriers. Courtesy of Carlos Cruchaga and Oskar Hariri.
This type of genomics work is in its infancy, but so far, it appears that people with dominantly inherited AD have fewer excitatory neurons and more neuronal death at the same disease stage than people with late-onset AD (see image above; Del-Aguila et al., 2019; Li et al., 2018). If this trend holds up, it would confirm at the level of gene expression that dominantly inherited Alzheimer’s disease is a more aggressive form of AD. “These are examples of the work we do with the tissue you donate,” Goate told the families.
Mutations Keep On Coming
Incidentally, pathogenic APP and presenilin mutations are popping up anew in people at a rate scientists can measure. While current DIAN members have (or had) a parent with the disease, separate collections of sporadic, i.e. non-familial, early onset AD cases turn out to contain people who carry known pathogenic mutations in APP or PSEN1 or 2. In Los Angeles, Goate cited a study that included 10 such cases in whom that known mutation had newly arisen, i.e., the patient’s biological parents had not had AD (Lanoiselée et al., 2017). “These mutations look sporadic now, but in future generations they will look familial,” Goate said. Luciana, an at-risk DIAN participant living in Argentina, showed such an example at the family conference. A journalist herself, she traced her family’s mutation to her Lebanese great-grandfather. He likely suffered what looked like sporadic early onset dementia in his day, but passed on a presenilin mutation to at least 10 descendants to date (see Part 1 of this series).
In fact, a growing number of APP and presenilin mutations—some familial, some sporadic—have become known in Central and South American countries in recent years. Jorge Llibre-Guerra at WashU told Alzforum that people afflicted with the A431E PSEN1 mutation, first discovered in the Mexican state of Jalisco, may turn out to be as numerous as those carrying the E280A “Paisa” mutation in Colombia, for whom Francisco Lopera and his team at University of Antioquia in Medellin have been providing care, support, and now a first prevention trial. At 1,192 mutation carriers, the Colombian cohort is the largest known in the world today.
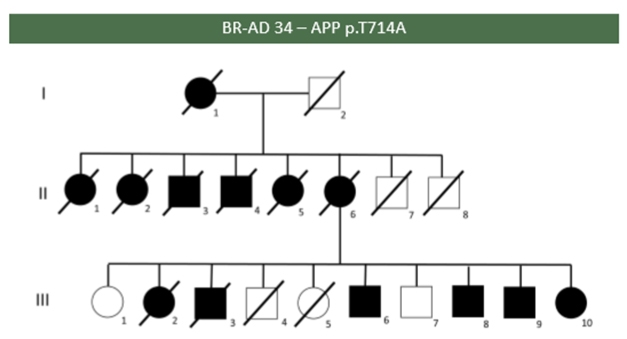
Affected (black) and unaffected members of a Brazilian family carrying the “Iranian” APP mutation T714A. Courtesy of Leonel Takada.
At the DIAD family meeting, Leonel Takada, University of São Paulo Medical School, showed a poster of the first Brazilian study assessing how frequently mutations can be found in autosomal-dominant AD at his clinic. Of the 17 probands from different families in Takada’s study, five had a mutation in PSEN1, of which four were previously described and one is new. Two probands from unrelated families had the same T714A APP mutation, known as the Iranian mutation. In one of the families, six of eight children of an affected mother had the disease, and the sixth sibling alone passed the mutation on to six of her 10 children (see image above).
Llibre-Guerra, Lopera, Takada, Ana Luisa Sosa-Ortiz of the National Institute of Neurology and Neurosurgery in Mexico City, and Ricardo Allegri of FLENI in Buenos Aires are working with the Alzheimer’s Association to obtain funding for a DIAN site to bring services, research, and eventually also prevention trials to families in this part of the world.
To learn about a genetic therapy for AD, see Part 3 of this series; for an update on ongoing and planned DIAN trials, see Part 4.—Gabrielle Strobel
References
Mutations Citations
News Citations
- The Mutation You Want: It Protects the Brain, Extends Life
- Genetic Resilience—Can We Find Treatments for the Sick by Studying the Healthy?
- ASOs: Wave of the Future in Alzheimer’s Therapeutics?
- As DIAN Wraps Up Anti-Aβ Drug Arms, it Sprouts Tau, Primary Prevention Arms
Paper Citations
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991 Feb 21;349(6311):704-6. PubMed.
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995 Jun 29;375(6534):754-60. PubMed.
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995 Aug 31;376(6543):775-8. PubMed.
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995 Aug 18;269(5226):973-7. PubMed.
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993 Aug 13;261(5123):921-3. PubMed.
- Campion D, Charbonnier C, Nicolas G. SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing data. Acta Neuropathol. 2019 Aug;138(2):173-186. Epub 2019 Mar 25 PubMed.
- Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, St Jean P, Lawson M, Ehm MG, Mayeux R, Goate AM, NIA-LOAD/NCRAD Family Study Consortium. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS One. 2012;7(2):e31039. Epub 2012 Feb 1 PubMed.
- Cruchaga C, Del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, Ibanez L, Deming Y, Kapoor M, Tosto G, Mayeux RP, Holtzman DM, Fagan AM, Morris JC, Bateman RJ, Goate AM, Dominantly Inherited Alzheimer Network (DIAN), Disease Neuroimaging Initiative (ADNI), NIA-LOAD family study, Harari O. Polygenic risk score of sporadic late-onset Alzheimer's disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement. 2018 Feb;14(2):205-214. Epub 2017 Sep 21 PubMed.
- Tavana JP, Rosene M, Jensen NO, Ridge PG, Kauwe JS, Karch CM. RAB10: an Alzheimer's disease resilience locus and potential drug target. Clin Interv Aging. 2019;14:73-79. Epub 2018 Dec 28 PubMed.
- Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, Ibanez L, Bradley J, Wang F, Bergmann K, Davenport R, Morris JC, Holtzman DM, Perrin RJ, Benitez BA, Dougherty J, Cruchaga C, Harari O. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther. 2019 Aug 9;11(1):71. PubMed.
- Li Z, Del-Aguila JL, Dube U, Budde J, Martinez R, Black K, Xiao Q, Cairns NJ, Dominantly Inherited Alzheimer Network (DIAN), Dougherty JD, Lee JM, Morris JC, Bateman RJ, Karch CM, Cruchaga C, Harari O. Genetic variants associated with Alzheimer's disease confer different cerebral cortex cell-type population structure. Genome Med. 2018 Jun 8;10(1):43. PubMed.
- Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauviré V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, Ceccaldi M, Félician O, Godefroy O, Formaglio M, Croisile B, Auriacombe S, Chamard L, Vincent JL, Sauvée M, Marelli-Tosi C, Gabelle A, Ozsancak C, Pariente J, Paquet C, Hannequin D, Campion D, collaborators of the CNR-MAJ project. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017 Mar;14(3):e1002270. Epub 2017 Mar 28 PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.