Peripheral Aβ Can Accumulate in Brain, Trigger Degeneration
Quick Links
Could Aβ from the blood contribute to Alzheimer’s disease? In the October 31 Molecular Psychiatry, researchers led by Yan-Jiang Wang at Daping Hospital, Third Military Medical University in Chongqing, China, and Weihong Song at the University of British Columbia, Vancouver, Canada, made a case for this. The researchers surgically joined AD model mice to wild-type mice such that the animals shared a blood supply, a technique known as parabiosis. Over several months, circulating human Aβ42 entered the brains of the wild-types. It accumulated there and formed plaques, though it did not appear to seed deposits with endogenous mouse Aβ. Plaque deposition in turn kicked off tau phosphorylation, inflammation, and degeneration.
- Wild-type mice shared a blood supply with AD mice for several months.
- The wild-types developed amyloid plaques and other signs of AD.
- This suggests peripheral Aβ could contribute to Alzheimer’s.
The finding implies that peripheral Aβ might contribute to the development of Alzheimer’s, the authors believe. “This suggests that periphery-derived Aβ would be an important target for the prevention and treatment of AD. Alzheimer’s might be a whole-body disease,” Song and Wang wrote to Alzforum.
Other researchers were intrigued, but said it is unclear whether the finding is clinically relevant. “This is an excellent paper,” Huntington Potter, director of the Rocky Mountain Alzheimer's Disease Center in Denver, wrote to Alzforum (see full comment below). “It’s an interesting model,” agreed Lary Walker at Emory University, Atlanta. Walker noted that, in people, the brain produces much more Aβ than does the periphery, implying that any contribution of peripheral peptide to disease would be small.
Researchers emphasized that people need not worry about contracting AD from blood transfusions. Konrad Beyreuther at the University of Heidelberg, Germany, and Colin Masters at the University of Melbourne, Australia, pointed out there is no evidence of Alzheimer’s or other neurodegenerative diseases being passed along in this manner (Edgren et al., 2016). “Single blood transfusions compared to 12 months [of] parabiosis are vastly different scenarios,” Beyreuther and Masters wrote to Alzforum (see full comment below).
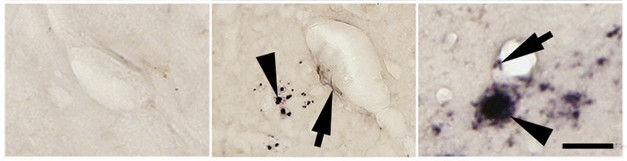
Aβ Infiltrates Brain. Unlike wild-type (left), parabiosis mice (middle) develop cerebral amyloid angiopathy (arrow) and amyloid plaques (arrowhead). The plaques are smaller than those in AD model mice (right). [Courtesy of Bu et al., Molecular Psychiatry.]
Previous research had found that peripheral Aβ can trigger plaque formation in AD mice, which overproduce the human peptide in the brain (Oct 2010 news; Jul 2014 news). In people, there is some evidence that peripheral Aβ can seed plaques. Researchers at University College London found amyloid plaques in the brains of young adults who had received intramuscular injections of growth hormone taken from the pituitary glands of elderly cadavers (Sep 2015 news). While these findings suggested that peripheral Aβ can enter the brain, it is unclear how big a role this route plays on its own.
Wang and colleagues set up their parabiosis experiments to distinguish the effect of peripheral Aβ from that of peptide made in the brain. First author Xian-Le Bu joined 10-month-old APPswe/PS1dE9 mice to age-matched wild-types, and analyzed how that changed the wild-type two, four, eight, and 12 months later. The scientists found human Aβ in the mouse brains starting at four months after parabiosis began. Insoluble Aβ aggregates appeared after eight months. After a year of parabiosis, the authors detected plaques in the walls of brain blood vessels and in the parenchyma surrounding the vessels. These plaques contained very little mouse Aβ, indicating that they were made almost entirely from circulating peptide. They were smaller and less extensive than the plaques found in transgenics (see image above).
Importantly, wild-type mouse brain exhibited other changes after a year of parabiosis. The amount of hyperphosphorylated tau climbed by 30 percent compared to that of age-matched controls. Activated astrocytes and microglia surrounded Aβ deposits, and levels of various pro-inflammatory cytokines roughly doubled, indicating neuroinflammation. About twice as many microhemorrhages developed as in age-matched controls. In addition, the authors detected more of the structural protein neurofilament 200 in the neurons of the neocortex, a sign of axonal degeneration.
Did these changes affect the mice’s function? The authors were unable to run behavioral experiments on the joined mice, but they measured long-term potentiation in hippocampal slices taken from them after four months of parabiosis. LTP in the parabiont wild-types was weaker than in age-matched controls, nearly matching the impairment in transgenic mice. This suggests peripheral Aβ can harm synapses, the authors noted.
This paper did not address whether peripheral Aβ was able to seed plaque formation in the brain by corrupting normal mouse protein. Nonetheless, commenters are curious about this. Beyreuther and Masters suggested trying parabiosis with APP knockout mice to determine whether the mouse peptide plays a role in plaque formation. Marco Colonna and Wilbur Song at Washington University in St. Louis wondered whether the circulating Aβ from the hAPP parabiont was produced in their periphery or the brain. “It would be interesting to see if the same phenomenon is observed with a knock-in AD model in which Aβ is only produced in brain,” they wrote (see full comment below).
Tony Wyss-Coray at Stanford University suggested that other blood-borne factors might have an effect as well. “It would be interesting to see to what extent inflammatory factors from the APP parabiont contributed to the amyloid and tau pathology in the recipient wild-type mouse,” he wrote to Alzforum (see full comment below).
Wang and Song believe that targeting peripheral Aβ might be a strategy for treating AD. They previously reported that lowering Aβ in the blood of mouse models reduced the peptide in the brain and improved memory (Xiang et al., 2015; Jin et al., 2017). They also note that people with cognitive impairment have been found to make more Aβ in platelets than do healthy people (Liu et al., 2007; Johnston et al., 2008). “We are investigating the peripheral pathogenesis of AD, and finding diagnostic biomarkers and developing therapies from this approach,” they wrote to Alzforum (Wang et al., 2017). The anti-Aβ antibody solanezumab, which fell short in Phase 3, was expected to lower plasma Aβ and thus draw it from brain, but stabilized levels in the blood (Jan 2017 news).—Madolyn Bowman Rogers
References
News Citations
- Peripheral Aβ Seeds CAA and Parenchymal Amyloidosis
- Does Aggregated Aβ Pass Directly From Blood to Brain?
- Alzheimer’s Transmission Between People? Amyloid Plaques in Hormone Recipients Hint at Prion-like Spread
- Solanezumab: Did Aβ ‘Reflux’ From Blood Confound Target Engagement in CSF?
Paper Citations
- Edgren G, Hjalgrim H, Rostgaard K, Lambert P, Wikman A, Norda R, Titlestad KE, Erikstrup C, Ullum H, Melbye M, Busch MP, Nyrén O. Transmission of Neurodegenerative Disorders Through Blood Transfusion: A Cohort Study. Ann Intern Med. 2016 Sep 6;165(5):316-24. Epub 2016 Jun 28 PubMed.
- Xiang Y, Bu XL, Liu YH, Zhu C, Shen LL, Jiao SS, Zhu XY, Giunta B, Tan J, Song WH, Zhou HD, Zhou XF, Wang YJ. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 2015 Oct;130(4):487-99. Epub 2015 Sep 12 PubMed.
- Jin WS, Shen LL, Bu XL, Zhang WW, Chen SH, Huang ZL, Xiong JX, Gao CY, Dong Z, He YN, Hu ZA, Zhou HD, Song W, Zhou XF, Wang YZ, Wang YJ. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017 Aug;134(2):207-220. Epub 2017 May 5 PubMed.
- Liu WW, Todd S, Craig D, Passmore AP, Coulson DT, Murphy S, Irvine GB, Johnston JA. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24(6):464-8. PubMed.
- Johnston JA, Liu WW, Coulson DT, Todd S, Murphy S, Brennan S, Foy CJ, Craig D, Irvine GB, Passmore AP. Platelet beta-secretase activity is increased in Alzheimer's disease. Neurobiol Aging. 2008 May;29(5):661-8. PubMed.
- Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017 Sep 29;13(10):612-623. PubMed.
External Citations
Further Reading
Primary Papers
- Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, Zhang K, Liu YH, Zeng F, Liu JH, Sun HL, Zhuang ZQ, Chen SH, Yao XQ, Giunta B, Shan YC, Tan J, Chen XW, Dong ZF, Zhou HD, Zhou XF, Song W, Wang YJ. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2017 Oct 31; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Heidelberg University
University of Melbourne
Bu et al. studied the transmission of blood-derived Aβ protein by using a model of parabiosis between APPswe/PS1dE9 transgenic AD mice and their wild-type littermates carrying the endogenous murine APP gene. In this way they made them share a blood system.
That brain entry of blood-derived human Aβ can occur was shown by two-photon intravital imaging after intravenous injection of 125I-labeled human Aβ42 in the brains of 10-month-old wild-type mice. After a 12-month period of parabiosis, human Aβ originating from APPswe/PS1dE9 transgenic had entered and formed cerebral amyloid angiopathy and human Aβ plaques in the brains of wild-type mice.
In addition, tau hyperphosphorylation, neurodegeneration, neuroinflammation, microhemorrhage and substantially impaired hippocampal LTP was also detected in the brains of the parabiotic wild-type mice. This strongly suggest that blood-derived Aβ can enter the brain, form the Aβ-related pathologic changes, and induce functional deficits of neurons.
These findings contradict a study by Edgren and his colleagues (Edgren et al., 2016) that tracked 1.47 million recipients of blood transfusions across Sweden and Denmark who received transfusions between 1968 and 2012. Of these, 2.9 percent received at least one transfusion from a donor diagnosed with a neurodegenerative disorder within 20 years after donation. The most common source was blood from donors with dementia of any type (1.8 percent), Parkinson’s disease (1.0 percent), Alzheimer’s disease (0.8 percent), and ALS (0.3 percent). No evidence of transfusion transmission of dementia, Alzheimer’s disease, or Parkinson’s disease was found considering disease concordance between donors and their respective recipients.
The discrepancy between evidence and absence of evidence of blood transmission of amyloid may be simply due to the induction of autoantibodies in humans who received blood from donors with amyloid. Autoantibody production against Aβ occurs frequently in the majority of human sera, as we showed some time ago (Mönning et al., 1990; Mönning et al., 1991). That the same did not occur in the model of parabiosis between APPswe/PS1dE9 transgenic AD mice and their wild-type littermates may simply be due to the high concentration of Aβ in blood of the APPswe/PS1dE9 mice. It is orders of magnitude higher than in human blood, allowing that blood-derived Aβ can enter the brain before induction and during the presence of autoantibodies.
Why does human Aβ enter and accumulate in the brains of the parabiotic wild-type mice? Obviously, they managed to get enough “seed” into the recipient brain by using parabiosis over a long period. There is no question murine Aβ can aggregate (Xu et al., 2015), it is just important to get enough seed into place. A straightforward test of this hypothesis would be parabiosis with APP ko mice.
A further consideration may be the form in which Aβ is delivered to the recepient: cell-free Aβ42 in plasma, exome-associated Aβ42 in plasma, or viable cellular elements (monocytes or brain-derived professional phagocytic cells carrying cargo of Aβ42). Single blood transfusions compared to 12-months parabiosis are vastly different scenarios.
References:
Edgren G, Hjalgrim H, Rostgaard K, Lambert P, Wikman A, Norda R, Titlestad KE, Erikstrup C, Ullum H, Melbye M, Busch MP, Nyrén O. Transmission of Neurodegenerative Disorders Through Blood Transfusion: A Cohort Study. Ann Intern Med. 2016 Sep 6;165(5):316-24. Epub 2016 Jun 28 PubMed.
Mönning U, Schreiter-Gasser U, Hilbich C, Bunke D, Prior R, Masters CL, Beyreuther K. Alzheimer amyloid BA4-protein reactive antibodies in human sera and CSF. Neurobiol Aging. (1990) 11, 338
Xu G, Ran Y, Fromholt SE, Fu C, Yachnis AT, Golde TE, Borchelt DR. Murine Aβ over-production produces diffuse and compact Alzheimer-type amyloid deposits. Acta Neuropathol Commun. 2015 Nov 14;3:72. PubMed.
Washington University School of Medicine
This is a very interesting paper. It clearly demonstrates that the wild-type partner of parabiosis is getting Aβ from the circulation somehow.
Given that this transgenic mouse model has exogenous Aβ expression, it is possible that circulating levels of Aβ are not physiological. Thus, one important question is which are the sources of Aβ in this AD mouse model and, in particular, whether there is any cell population in the periphery that is producing Aβ when it normally does not.
It would be interesting to see if the same phenomenon is observed with a knock-in AD model, in which Aβ is only produced in brain. It would also be interesting to see whether the same result can be obtained by simply injecting Aβ in the circulation intravenously.
Stanford University Medical School
The results in this paper are interesting and possibly consistent with the seeding findings by Jucker and others.
It would be interesting to see to what extent other (inflammatory) factors from the APP parabiont contribute to the amyloid and tau pathology in the recipient wild-type mouse. Also, I’d like to see mass spec analysis of the amyloid deposits to confirm their identity, and to see if they also contain mouse Aβ and whether they would form in the absence of mouse APP (similar to experiments done with Prp).
It is somewhat unclear to me if the finding is clinically relevant. Young people are obviously not exposed to aged plasma from AD patients, and it is unclear where the Aβ seeds would come from. Having said that, it might be interesting to express human mutant APP in systemic organs and see if the same pathology develops. After all, APP is expressed in many tissues.
University of Colorado Alzheimer’s and Cognition Center
This is an excellent paper. Well-performed and -analyzed experiments clearly show that peripheral Aβ, derived from an APP/PS transgenic mouse, can be delivered through the circulation by parabiosis to the blood and brain of a recipient wt mouse, where it can generate Aβ-containing amyloid plaques and CAA, as well as hyperphosphorylated tau and other features of AD pathology and also lead to reduced LTP.
The papers adds importantly to our (Nilsson et al., 2012) and Wyss-Coray and colleagues’ previous studies of parabiosis to study Alzheimer’s disease and aging, respectively. Curiously, we found that parabiosis did not allow ApoE, an essential catalyst of amyloid formation, to enter the brain and induce amyloid formation in a recipient APP/PS transgenic animal expressing Aβ but no ApoE. Thus the availability of Aβ and ApoE from the circulation to affect amyloid formation in the brain are different, with implications for therapy.
References:
Nilsson LN, Gografe S, Costa DA, Hughes T, Dressler D, Potter H. USE OF FUSED CIRCULATIONS TO INVESTIGATE THE ROLE OF APOLIPOPROTEIN E AS AMYLOID CATALYST AND PERIPHERAL SINK IN ALZHEIMER'S DISEASE. Technol Innov. 2012 Feb 1;14(2):199-208. PubMed.
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011 Sep 1;477(7362):90-4. PubMed.
RIKEN Center for Brain Science
Parabiosis causes a number of extreme immunological responses. The weakness of this study is that there are no relevant negative controls.
Icahn School of Medicine at Mount Sinai
This study is elegant and intriguing. It appropriately takes advantage of several sophisticated methodologies to address a technically difficult concept.
While the authors show that human Aβ from the transgenic model does appear in the brains of connected WT mice, it is unclear whether the associated pathologies the authors assessed many months after parabiosis are a direct result of the penetrating blood-derived Aβ or simply a result of the overexpression in blood. In other words, overexpression of human Aβ in the transgenic mouse likely alters some portion of the shared plasma proteome in a way that could conceivably alter the partner WT brain and its development of tau pathology, microgliosis, etc.
Along a similar “vein,” do these putative changes in blood alter the neurovascular unit in a way that leads to these pathologies or shapes LTP independent of amyloid? If indirect pathways are not suspected, is the Aβ entry facilitated by receptors like RAGE (Deane et al., 2003), or does the penetration occur via passive routes? The kinetics shown in Figure 1 suggest otherwise, but this could be an important direction to pursue if indeed the accumulation from blood is substantial even when repeated in APP knock-in models.
Finally, the accumulation of blood-derived human Aβ appears to be most dramatic in aged mice (22 months) that have shared transgenic blood for 12 months—an age at which some have argued that blood-brain barrier disruption occurs, though this has been disputed in some models (Bien-Ly et al., 2015). Possible disruption in the neurovasculature in the aged parabiont brain could presumably allow Aβ and its seeds to penetrate the parenchyma in a way that may not occur in the aged human brain.
References:
Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W, Shihadeh V, Ulufatu S, Foreman O, Lu Y, DeVoss J, van der Brug M, Watts RJ. Lack of Widespread BBB Disruption in Alzheimer's Disease Models: Focus on Therapeutic Antibodies. Neuron. 2015 Oct 21;88(2):289-97. PubMed.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003 Jul;9(7):907-13. PubMed.
Daping Hospital
We would like to thank all for their comments. With regard to Joseph Castellano’s comment in particular: It is reasonable that certain substances in the blood of AD mice, such as inflammatory cytokines, may enter the blood of parabiotic wild-type mice, and promote AD-associated pathologies such as neuroinflammation, tau hyperphosphorylation and overexpression of endogenous mouse Aβ in the brain. However, in our study the most important pathologies, which are the neuritic plaques and CAA formed in brains of parabiotic mice, are mainly composed of human Aβ. And the activated glial cells are also presented adjacent to human Aβ deposits.
These findings suggest that AD-associated pathologies and neuronal dysfunctions observed in the brains of parabiotic wild-type mice are most likely the direct results of penetration of human Aβ from blood into the brain. We agree that the accumulation of human Aβ in brains of aged parabiotic wild-type mice should be due to the compromised Aβ clearance capacity and the disrupted neurovasculature during aging.
Panjab University
I am curious to know:
UCC Medical School
The work presented here directly confirms that systemic Aβ peptide from the circulation is involved in Alzheimer’s development.
The question now arises if the peptide was generated (i) in the brains of the transgenic animals, then appeared in the circulation and after that penetrated to the brains of wild-type, or (ii) in the blood of the transgenic animals and just passed to the brains of wild-type animals.
We recently published research showing massive release of Aβ peptide during experimental thrombosis, with platelets being the source (Kucheryavykh et al., 2017). My colleagues and I are convinced that platelets are the main source of Aβ peptide, causing neuronal damage as well. We also suggested that Aβ is an unrecognized natural antibiotic, released in many septic and aseptic causes. We reviewed indirect research studies that confirm Aβ generated in blood as a local response to the inflammation/antigen may penetrate the brain barrier to add to the Alzheimer’s disease burden (Inyushin et al., 2017). In this aspect, the discussed work is a brilliant confirmation that Alzheimer’s is a blood-associated disease.
References:
Kucheryavykh LY, Dávila-Rodríguez J, Rivera-Aponte DE, Zueva LV, Washington AV, Sanabria P, Inyushin MY. Platelets are responsible for the accumulation of β-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res Bull. 2017 Jan;128:98-105. Epub 2016 Nov 28 PubMed.
Inyushin MY, Sanabria P, Rojas L, Kucheryavykh Y, Kucheryavykh L. A β Peptide Originated from Platelets Promises New Strategy in Anti-Alzheimer's Drug Development. Biomed Res Int. 2017;2017:3948360. Epub 2017 Sep 5 PubMed.
Make a Comment
To make a comment you must login or register.