Network Analysis Points to Distinct Effects of Amyloid, Tau
Quick Links
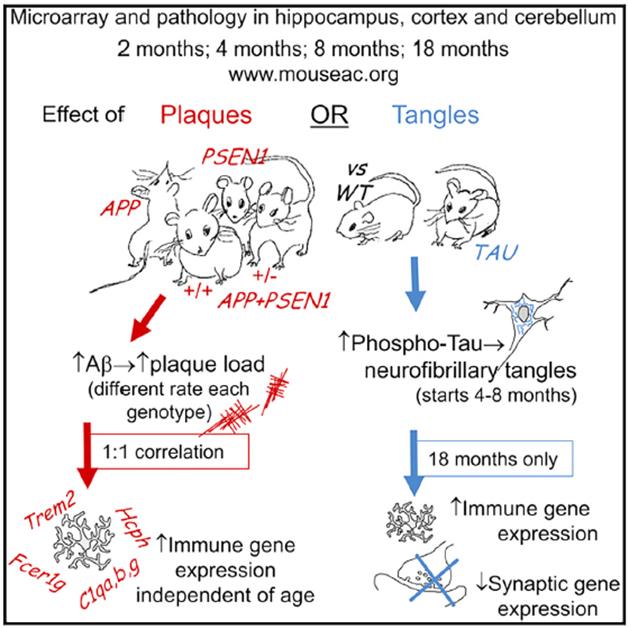
Amyloid plaques and tau tangles both characterize Alzheimer’s, but how does each drive the disease? A new study in the February 3 Cell Reports aims to disentangle their relative contributions to AD pathology. Researchers led by Frances Edwards and John Hardy at University College London analyzed global gene expression in the cortex and hippocampus at different ages in several AD mouse models that develop either plaques alone or tangles alone. Amyloid plaques triggered immediate and dramatic neuroinflammation, but did not affect synaptic genes, the authors report. In tau models, inflammation lagged several months behind the appearance of tangles, while expression of synaptic proteins plummeted in the late stages of disease.
Both pathologies boosted many of the same immune genes, including several known AD risk factors, such as TREM2. In addition, network analysis flagged similar key genes as drivers of the immune response in both types of mice, although some intriguing differences between the models emerged as well. The dataset is publicly available online, and could provide a useful resource to other investigators, the authors suggest.
Commentators agreed, praising the depth and thoroughness of the investigation. “This was much more comprehensive than other [mouse] studies I have seen, and allows for the comparison of different models, which is critical,” said Michael Sasner, who directs bioinformatics and model development at the Jackson Lab, Bar Harbor, Maine. Gerold Schmitt-Ulms at the University of Toronto wrote to Alzforum, “As a researcher undertaking conceptually similar proteomics analyses, I certainly look forward to using this resource to look up levels of gene products which we flagged in our own AD data sets.” (See full comment below.) Greg Cole at the University of California, Los Angeles, noted, “It’s the first time I’ve seen someone do a network analysis [in mice] similar to what has been done in humans.”
Previously, researchers led by Valur Emilsson at the Icelandic Heart Association and Eric Schadt at Icahn School of Medicine at Mount Sinai, New York, reported that networks of immune genes go haywire in AD patients (see May 2013 webinar), while an analysis of human data by Mina Ryten and colleagues, including Hardy, at UCL likewise highlighted the linchpin role TREM2 plays in the brain’s response to Alzheimer’s (see Forabosco et al., 2013). In animal work, many studies have catalogued soaring levels of immune gene expression in AD mouse models (see, e.g., May 2004 news; Wu et al., 2006; Wirz et al., 2013), but no study had systematically compared amyloid to tau models.
Amyloid vs. Tau. Amyloid mice develop inflammation, whereas tau mice lose synapses, according to gene-expression analyses of several models. [Image courtesy of Cell Reports, Matarin et al., 2015.]
Edwards and colleagues collaborated with the pharmaceutical company GlaxoSmithKline in Stevenage, U.K., to analyze five of the company’s mouse models. The TAS10 model overexpresses APP with the Swedish mutation, while the TPM strain overexpresses human mutant presenilin 1. Crossing these strains yields heterozygous TASTPM hybrids; these mice can be bred to homozygosity at both alleles (TASTPM HO), resulting in more aggressive pathology. The fifth model, TAU, expresses human tau with the P301L mutation, driven by the CaMK-II promoter, and develops neurofibrillary tangles and neurodegeneration.
First author Mar Matarin compared microarrays of global gene expression from the hippocampus, cortex, and cerebellum from all five models and age-matched wild-type controls. The authors collected data from 2-, 4-, 8-, and 18-month-old mice, uncovering distinct early and late effects. At the youngest ages, both the amyloid and tau models had altered levels of synaptic genes in the hippocampus compared to controls, with some genes up and others down. Genes involved in calcium signaling were suppressed in the hippocampus in both amyloid and tau mice, in line with other studies fingering calcium dysregulation as an early culprit in AD (see, e.g., Dec 2014 conference news; May 2014 news; April 2014 news). These synaptic and calcium disruptions normalized to control levels by 8 months of age, however.
A different picture emerged in aged mice, in which immune gene expression ran rampant in the cortex and hippocampus of most models. Few genes changed in the cerebellum, which is largely spared in AD. The four amyloid models provided a varying time course of plaque development, with homozygous TASTPM animals accumulating plaques as early as 4 months, heterozygotes at 8 months, TAS10s at 18 months, and TPMs never getting them. The authors correlated plaque presence with the explosion of immune genes and found nearly a 1:1 relationship. “This was a surprise,” Edwards told Alzforum. “The increase in immune gene expression goes hand-in-hand with plaques. It emphasizes that the immune response plays a key role in Alzheimer’s.” What remains unclear is whether this immune response helps the organism or does further harm, she added.
By contrast, the tau mice developed tangles at 8 months old, but immune genes did not peak until 18 months. At that age, synaptic gene expression crashed, while some cell-death genes rose. These changes likely reflect ongoing neurodegeneration and loss of synapses, the authors suggest. The results jibe with other studies linking tau pathology to synaptic loss and cognitive decline (see Nov 2013 conference news; Aug 2014 conference news).
Drilling down to specific genes, the authors found several AD risk factors elevated in both elderly tau and homozygous APP/PS1 (TASTPM) mice. These included TREM2, CD33, INPP5D, MS4A6D, and ApoE. TREM2 was the most amplified, up more than fivefold in the aged homozygous TASTPM mice.
The researchers then analyzed relationships between overexpressed immune genes to find those that correlated most strongly with the largest numbers of other genes. This network analysis pointed to just a handful of “hub genes” in amyloid mice: TREM2, C1q, FCER1G, and HCPH in the hippocampus, and TREM2, C1q, FCER1G, and TLR2 in the cortex. Similar hub genes cropped up in the hippocampus of tau mice (C1q, FCER1G, and HCPH). “It’s quite remarkable how similar the immune system hub genes are between different brain regions and models,” Edwards noted. On the other hand, the cortical network in tau mice was distinct, having only two genes (C1q and TLR2) in common with the others. Frontotemporal dementia, a common tauopathy modeled by the tau mice, mainly perturbs cortical cognitive processes.
What do these findings mean? Hub genes likely drive changes in other genes, Edwards said. TREM2 marks activated microglia, and its status as a hub gene in amyloid mice suggests that microglia activate in reaction to plaques. In tau mice, although TREM2 is elevated, it falls short of hub status. In these animals, microglia may play a less prominent role, Edwards suggested. Cole speculated that the inflammatory response in tau mice might relate more to neurodegeneration, rather than glial changes as in amyloid mice.
The roles of other hub genes are less clear. TLR2 (toll-like receptor 2) is a microglial receptor with a similar function to TREM2 that can interact with the latter (see Paradowska-Gorycka and Jurkowska, 2013). The complement protein C1q has become a focus of AD research. It not only mediates phagocytosis of debris, but also pruning of synapses during development (see Nov 2007 conference news), and previously had been shown to turn on in response to both plaques and tangles (see Nov 1998 news). FCER1G codes for a subunit of an IgE Fc receptor, while HCPH codes for a phosphatase predominantly found in hematopoietic cell lines.
In future work, Edwards will manipulate levels of various hub genes in these mouse models and examine the effects on pathology in order to glean clues to the function of each gene. She will also do a comparative-expression analysis in an APP knock-in mouse model (see April 2014 webinar), which expresses endogenous levels of protein. “If the data come out similarly, it will really support that these are the genes that matter,” Edwards said.
Sasner suggested that it would be valuable to do similar gene expression analyses on other models, particularly one that includes both amyloid and tau pathology. The comparison might reveal an additive or synergistic effect from having both pathologies, he noted. The approach could also help validate animal models relative to patient populations, he added.
Piet Eikelenboom at Vrije University, Amsterdam, noted that the amyloid mice probably model the early stage of Alzheimer’s pathology, which is dominated by plaque deposition but few tangles and little degeneration. “I found it very surprising to see in these models the interesting data about the involvement of immune system in the early stages of AD pathology. This fits with our findings obtained from human AD brains (see Hoozemans et al., 2006). These transgenic models seem very promising for deciphering and manipulating these immune system factors,” he wrote to Alzforum.—Madolyn Bowman Rogers
References
Webinar Citations
- Can Network Analysis Identify Pathological Pathways in Alzheimer’s
- Good-Bye Overexpression, Hello APP Knock-in. A Better Model?
News Citations
- GLOing Reports from Gene Profiling of Mouse Models
- Calcium Disruptions Wreak Synaptic Havoc
- Inositol Triphosphate Receptor Blamed for Calcium Chaos in Alzheimer’s
- Calcium Sensor STIM2 Maintains Synapses, Ebbs in Alzheimer’s
- Do Tau Tracers Track Cognitive Decline in Disease?
- San Diego: MHC Class I and Complement—Holding Down Second Jobs in the Synapse
- 1998 Society for Neuroscience Meeting: Complement Cascade Activation by Neurofibrillary Tangles
Research Models Citations
Paper Citations
- Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, Weale ME, Ryten M. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 Dec;34(12):2699-714. PubMed.
- Wu ZL, Ciallella JR, Flood DG, O'Kane TM, Bozyczko-Coyne D, Savage MJ. Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease. Neurobiol Aging. 2006 Mar;27(3):377-86. PubMed.
- Wirz KT, Bossers K, Stargardt A, Kamphuis W, Swaab DF, Hol EM, Verhaagen J. Cortical beta amyloid protein triggers an immune response, but no synaptic changes in the APPswe/PS1dE9 Alzheimer's disease mouse model. Neurobiol Aging. 2013 May;34(5):1328-42. PubMed.
- Paradowska-Gorycka A, Jurkowska M. Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum Immunol. 2013 Jun;74(6):730-7. Epub 2013 Feb 28 PubMed.
- Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P. Neuroinflammation and regeneration in the early stages of Alzheimer's disease pathology. Int J Dev Neurosci. 2006 Apr-May;24(2-3):157-65. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS, Perona M, Desai R, Smith KJ, Latcham J, Fulleylove M, Richardson JC, Hardy J, Edwards FA. A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 2015 Feb 3;10(4):633-44. Epub 2015 Jan 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Toronto
This study by Matarin et al. is, by a considerable margin, the largest microarray-based investigation of Alzheimer’s disease (AD) mouse models to date. The authors monitored and compared transcript levels of more than 12,500 genes in five AD models (plus one control mouse line) at four different ages and for three different brain regions. To my knowledge, the scale of this study is only eclipsed by one prior herculean AD-related microarray investigation, which compared gene expression patterns in three brain regions of 600 human brains (Podtelezhnikov et al., 2011). Both studies were complementary in their design and pointed toward a distortion of immune or inflammatory response gene products early in the disease.
This kind of discovery research is urgently needed and incredibly useful, yet is also renowned for falling short on follow-up data analysis and validation, making it difficult for the reader to extract useful information. The authors avoided this trap by following their microarray tour de force with a careful bioinformatic evaluation. In particular, the network analysis of immune genes and the comparison of microarray and GWAS data sets added value and revealed useful insights. From my perspective, two interesting observations stood out:
1. Whereas extracellular amyloid disease primarily underlies inflammatory perturbations, the deposition of tau correlates better with changes to transcripts coding for synaptic proteins. The relationship of Aβ and tau pathologies to synaptic deficits is a matter of considerable contention in the field. It is refreshing to learn that data from a large-scale discovery approach place the blame for synaptic deficits squarely on tau. This finding contributes to understanding the basis of distinct presentations of AD and FTD-17—as also noted by the authors.
2. Trem2, one of the stronger risk factors for developing late-onset AD (Guerreiro et al., 2013; Jonsson et al., 2013), may steer changes to the immune response downstream of amyloid pathology but may play a lesser role in the brain’s response to tau deposition. The proposition that a subset of the new AD risk genes that emerged in large-cohort GWAS studies, including Trem2, influence Aβ rather than tau biology is not new, but the current study makes a particularly compelling case in its favor due to the rigorous, hypothesis-free approach it pursued.
As with any study, there are also caveats with this work. For example, the overexpression of APP transgenes in three of the four amyloid AD mouse models investigated represents a shortcoming accentuated by the heightened awareness that non-physiological levels of non-Aβ products of APP can skew phenotypes (Saito et al., 2014). Moreover, although the study is suggestive of a scenario whereby synaptic deficits in AD are predominantly driven by tau pathology, this conclusion has to remain tentative. The systems biology that precipitates synaptic deficits may be sufficiently different when both Aβ and tau etiologies unfold together, an aspect of AD not modeled in the current study. To begin to address this question, it would be interesting to learn about differences and commonalities in the data of this and the aforementioned human AD microarray study.
It is safe to assume that the extensive inventory of AD-related gene expression data will serve as a valuable resource that may unleash its full potential only in the years to come. As a researcher undertaking conceptually similar proteomics analyses, I certainly look forward to using this resource to look up levels of gene products which we flagged in our own AD data sets.
References:
Podtelezhnikov AA, Tanis KQ, Nebozhyn M, Ray WJ, Stone DJ, Loboda AP. Molecular insights into the pathogenesis of Alzheimer's disease and its relationship to normal aging. PLoS One. 2011;6(12):e29610. PubMed.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):117-27. Epub 2012 Nov 14 PubMed.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):107-16. Epub 2012 Nov 14 PubMed.
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014 May;17(5):661-3. Epub 2014 Apr 13 PubMed.
View all comments by Gerold Schmitt-UlmsUniversity College London
Thank you for your comment. I completely agree with the caveat concerning the overexpression of APP and PSEN1. We are now breeding the APPKI mice from Takaomi Saido’s group (see Saito et al., 2014) and are fully intending to add the data on these mice to mouseac.org as they come through. The comparison will be very interesting indeed. Where they show similar effects it will confirm the interpretation, but where they are different will add additional information about the role of APP itself. It is interesting to note, however, that whether heterozygous or homozygous, and whether with or without the additional PSEN1 transgene, the correlation of the immune gene expression to plaques is very close to 1:1, suggesting that it is indeed the plaques that are in the driver’s seat.
References:
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014 May;17(5):661-3. Epub 2014 Apr 13 PubMed.
View all comments by Frances EdwardsDeceased, 2016
In this manuscript, Matarin and colleagues show, for the first time, a comparison of changes in mRNA expression with progressive pathology in different transgenic mouse models of Alzheimer’s disease. Analyses of the hippocampus, cortex, and cerebellum were performed in five mice; four driven by mutant APP and/or PS1 transgenes on thy1 promoters, and one driven by mutant tau transgene overexpression on the CaMKII promoter. This manuscript shows that the accumulation of plaques very tightly associated with an increase in expression of genes associated with immune and inflammatory responses. On the other hand, when tau was overexpressed in forebrain neurons, there was a down-regulation of genes associated with synaptic function and neurotransmitter release and transport.
The authors also analyzed how the expression of AD risk genes previously identified by GWAS changed in these mice in order to help elucidate the value of these models of disease. Notably, changes in expression of Apoe, CD33, Inpp5d, Ms4a6d, and Trem2, were observed in some of the models, particularly in the tau and APP+PS1 lines, suggesting that these transgenic mice do in fact appropriately model at least some aspects of AD. The authors conclude from this study that targeting the accumulation of tau pathology would be most effective in abrogating the progression of memory decline associated with late stage AD pathology, since it was closely tied to suppressed expression of components vital to synaptic function, while targeting amyloid pathology could decrease activation of the immune system. These findings lend further support to the idea that Aß triggers the dysfunction in the brain, perhaps through immune activation, while tau accumulation is ultimately responsible for the neurodegeneration that leads to late-stage decline. Moreover, the authors have provided a comprehensive database that can now be used by any lab to further investigate gene expression changes in these mouse models. This database is a valuable tool for anyone working in these models.
View all comments by Chad DickeyVrije Universiteit
VU University Medical Center
The reported finding of increased immune gene expression as an early event in transgenic Alzheimer’s disease (AD) mice models is, for several reasons, very intriguing:
1. A major concern—frequently raised in connection with transgenic AD mice models harboring causal mutations—is their relevance to sporadic late-onset AD, which is by far the most common form of the disease. In this respect, it is noteworthy that the reported immune gene expression changes in the Aβ mice pertain to genes coding for complement factor C1q and microglia receptors, which can already be found in early stages of AD pathology in the brain. Immunohistochemical studies show that diffuse Aβ plaques, the initial neuropathological lesion, are immunolabeled for the early complement factors and that upregulation of monocyte/macrophage receptors on glia cells is seen at early stages of AD pathology, before the appearance of tau-related neuropathological changes (Eikelenboom et al., 2011). Furthermore, it is striking that the reported hub genes for early immune factors in the transgenic AD mouse models are related to genes that are genetic risk factors for the late-onset form of AD. It is intriguing to see the similarities between the early changes in the immune gene expression in transgenic AD mice models, and the genetic risk factors and pathological findings for related immune changes in the early pathological stages of AD.
2. Matarin and co-workers discussed that their findings indicate a separate involvement of the immune system in the early and late stages of AD pathology. We found evidence for upregulation of inflammatory mediators, complement proteins, and adhesion molecules in the initial stages of AD pathology. The early brain changes associated with neuroinflammation precede the extensive process of tau pathology related to neurodegeneration (Hoozemans et al., 2006). This early neuroinflammatory response closely associates with an aberrant regenerative response that has been well studied in the past by Carl Cotman (Irvine Institute) and Thomas Arendt (Leipzig). These findings in human AD brains are in line with the findings of Matarin, which suggest that the late but not the early immune changes are closely related with the Aβ-induced neurotoxicity.
3. Network analysis shows that upregulated gene expression of complement protein C1q belongs to the hub genes in the different models. Immunohistochemical studies of the AD brain have shown that amyloid plaques contain the early complement factors C1q, C4, and C3, as well as late complement factors including the lytic membrane attack complex. It is generally thought that these complement factors are involved in fibrillary Aβ-induced neurotoxicity. However, the early complement factors already can be detected in earliest stages of AD brain pathology, the diffuse plaques, while the late complement proteins of the membrane complement factor are found in later stages of AD pathology and only in mature plaques with tau positive neurites (Eikelenboom et al., 2006). So, the complement proteins can play a distinct role in the different stages of the pathological cascade in AD brains. In initial stages, the earlier complement factors could be involved in the process of abnormal synapse modulation, while in later stages the full complement activation, including formation of the membrane attack complex, could be involved in Aβ-induced neurotoxicity. This interpretation is in agreement with the findings of Matarin et al., indicating that in the transgenic AD mice models the late, but not the early, immune changes are related to synapse and neuronal loss.
In conclusion, the transgenic AD mice are interesting models in which to investigate the interactions between immune system-related factors and Aβ deposition in the early pathological stages of AD. Moreover, the transgenic AD models also seem relevant and promising for a better understanding of the role of inflammation in the late-onset form of AD.
References:
Eikelenboom P, Veerhuis R, van Exel E, Hoozemans JJ, Rozemuller AJ, Van Gool WA. The early involvement of the innate immunity in the pathogenesis of late-onset Alzheimer's disease: neuropathological, epidemiological and genetic evidence. Curr Alzheimer Res. 2011 Mar;8(2):142-50. PubMed.
Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P. Neuroinflammation and regeneration in the early stages of Alzheimer's disease pathology. Int J Dev Neurosci. 2006 Apr-May;24(2-3):157-65. PubMed.
Eikelenboom P, Veerhuis R, Scheper W, Rozemuller AJ, Van Gool WA, Hoozemans JJ. The significance of neuroinflammation in understanding Alzheimer's disease. J Neural Transm. 2006 Nov;113(11):1685-95. PubMed.
View all comments by Jeroen HoozemansUniversity College London
We thank Dr. Eikelenboom and Dr. Hoozemans for their comment and for pointing out highly relevant studies and reviews confirming that the mouse and human data are so consistent. The different roles of the immune system at different stages of the disease and in relation to plaques versus neurofibrillary tangles will be key to understanding what approaches can be taken for therapy as the disease progresses. This complexity underlines the importance of carefully matching therapeutic agents to the stage of disease progression where they may be effective when planning clinical trials. It is highly likely that an agent that prevents or delays the progression of the early phases of Alzheimer's disease will be ineffective or even damaging at later stages. Poor timing has very likely been an important element in the failure of previous clinical trials and may be particularly important where manipulation of the immune system is concerned.
View all comments by Frances EdwardsMake a Comment
To make a comment you must login or register.