From Near and Far, Aβ Beckons Tau to Tangle in the Cortex
Quick Links
Just as gravity explains how two distant objects can sway each other’s movement in space, synaptic connections may explain how amyloid plaques coax distant tau tangles to spread in the brain. This is according to a study published in Neuron on April 19 and led by Joon-Kyung Seong at Korea University in Seoul and William Seeley at the University of California, San Francisco. The scientists report that synaptic connections between multiple neocortical areas, where amyloid aggregates, somehow entice tau tangles to go beyond the entorhinal cortex, where they first form. Then, tangles spread into connected, nearby regions in the medial temporal lobe. Later, once tangles push even farther, into neurons within the inferior temporal gyrus, the two pathologies mingle for the first time, and it is this direct encounter that accelerates propagation and spread of tangles throughout the brain. The findings suggest that a one-two punch of remote and local interactions between Aβ and tau link the two pathological hallmarks of AD, and that amyloid-targeted therapies will have the greatest impact before plaques and tangles meet “face to face.”
- Long-distance connections between tangle-bearing neurons and Aβ-burdened regions coax initial tau spreading.
- Direct meeting of Aβ and tau in the inferior temporal gyrus sparks acceleration of tauopathy.
- In preclinical AD, tau tangles can spread via different routes.
“The study is a major step forward in understanding the interaction between Aβ and tau pathology, which emphasizes the critical role of brain connectivity in the development of AD,” wrote Nicolai Franzmeier of Ludwig-Maximilians University in Munich, Oskar Hansson and Rik Ossenkoppele of Lund University in Sweden, and Jacob Vogel of McGill University in Montreal, in a joint comment to Alzforum.
But it doesn't happen the same way in every person. While these findings explain the most commonly observed path of tangle progression in the brain—i.e., Braak's stages going from the medial temporal lobe (mTL) out into the neocortex—scientists also see alternate paths in some people. On April 18 in JAMA Neurology, Elizabeth Mormino at Stanford University and colleagues report that, at the preclinical stage of AD, about one in 10 people accumulate substantial tangles outside of the mTL. Curiously, they had no memory deficits, which usually accompany spread of tau beyond the mTL. On March 31 in Brain Communications, Tengfei Guo at the Shenzhen Bay Laboratory in China reported that in cognitively normal people whose amyloid PET scan is positive but CSF Aβ is normal, tangles accumulate broadly in the cortex, not the mTL. In all, the findings from all three papers hint that a person’s structural connectome of the brain, as well as Aβ accumulation patterns, may steer the path of their tau tangles as they develop Alzheimer’s disease.
A long-standing question in the field is how Aβ amyloidosis fuels the devastating spread of tau tangles throughout the brain. Amyloidosis first ignites in the neocortex, and is widely disseminated there before tau tangles creep out of the entorhinal cortex and into nearby temporal cortices. Despite this spatial separation of the two pathologies, multiple studies suggest that plaque accumulation in the neocortex is a requisite for the spread of tangles out of the EC, an event that heralds neurodegeneration and cognitive impairment (Mar 2016 news; Feb 2018 news; Adams et al., 2019).
First author Wha Jin Lee from Korea University and colleagues investigated this conundrum using a combination of amyloid-PET, tau-PET, and structural brain connectivity data. Before investigating how amyloid influenced tau’s trajectory, they mapped how tau tangles proceed through the brain by examining their distribution on tau-PET scans from 283 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), who spanned the clinical AD spectrum. In so doing, they approximated a “pseudo-longitudinal” trajectory of neurofibrillary tangle pathology. As expected, this identified the entorhinal cortex as the region to accumulate tangles first. Tangles were largely confined to this region in the preclinical stage of AD, and the brain-wide tau-PET signal was dramatically higher in people with late mild cognitive impairment (MCI).
To probe how neural circuitry plays into the propagation of tau tangles, the scientists used diffusion tensor imaging to construct a standard brain connectome, i.e., a matrix that describes how strongly each brain region is connected to each of 213 others. Integrating this matrix with the tau trajectory, the researchers modeled the most likely connections that explain tau’s spread. One region—the inferior temporal gyrus—stood out as a propagation hub. Seeley likened the ITG to a train depot in a big city. The ITG had myriad connections with regions that would subsequently develop tau tangles, and tau’s arrival at this hub heralded the acceleration phase of neurofibrillary tangle progression.
This route of tangle progression observed in the ADNI cohort matched up with data from a second, independent cohort in from Gangnam Severance Hospital in Korea, which included 304 participants spanning the clinical spectrum of AD.
So how does amyloid fit in? To gauge how plaques affect this progression, the researchers calculated an “amyloid influence metric” for each of the 213 regions. It reflects how strongly each region connects to regions with amyloid plaques. Multiplying a region’s amyloid metric by its tangle burden offered a measure of remote Aβ-tau interactions. Intriguingly, the entorhinal cortex scored higher than any other region: Not only did it have abundant tau tangles, it was also tightly connected to multiple brain regions burdened by plaques.
The researchers next surveyed amyloid and tau PET scans for regions where Aβ plaques directly interact with tangle-bearing neurons. The ITG scored higher than any other brain region in this regard, because both Aβ plaques and tangles were abundant there. Similar findings from the Korean cohort supported the idea that a combination of remote and local interactions between Aβ and tau pathologies may steer the pathophysiological progression of AD.
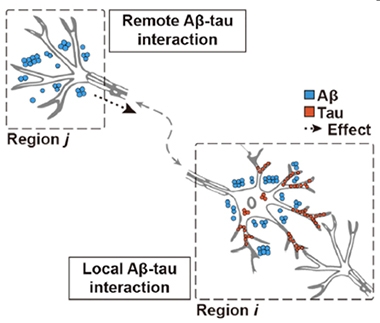
Long Distance and Local. Remote connections between regions with tangles (Region i) and regions with plaques (Region j) define the remote “amyloid influence metric.” Aβ and tau may also physically co-mingle, as in region i. [Courtesy of Lee et al., Neuron, 2022.]
While these findings were pieced together using cross-sectional data, a subset of participants in both the ADNI and Korean cohorts had serial PET scans. This longitudinal data taught the researchers that people whose amyloid influence score in their EC was high at baseline were likelier to have tau spread beyond the EC into nearby connected regions on follow-up. Additionally, those who had both Aβ plaques and tau tangles in the ITG at follow-up had much more widespread tangle pathology throughout the brain than did people who only had plaques in the ITG at follow-up.
By combining longitudinal and cross-sectional data from both cohorts, the researchers pieced together a model of tangle accumulation over time, marked by two transition points. The first occurs when amyloid plaques accumulate in multiple neocortical areas that connect with tangle-bearing neurons in the EC. Something about this remote Aβ-tau interaction triggers tangles to invade nearby, connected regions of the hippocampus, amygdala, and temporal cortices. The second pivotal moment occurs when tau tangles and Aβ plaques convene in the ITG. This toxic duo catalyzes widespread tau propagation into the ITG’s many connected regions, in what the researchers called the “acceleration phase” of AD (see image below).

Go Forth and Multiply. In the AD progression model, tangles begin in the lateral entorhinal cortex (red dot). Synaptic connections between plaque-ridden regions in the neocortex (blue dots) drive tangles beyond the EC. Later, local interactions between plaques and tangle-laden neurons in the ITG (purple dot) herald the acceleration phase of tau propagation across the neocortex. [Courtesy of Lee et al., Neuron, 2022.]
“The study is of high interest for the field, as the authors propose an integrated framework for how Aβ and tau pathology may interact remotely via neuronal connections as well as locally to give rise to the brain-wide spreading pattern of tau pathology that is seen in AD,” wrote Franzmeier and colleagues. They also noted important questions, including what molecular and cellular mechanisms underpin this Aβ-fueled tauopathy.
Seeley thinks the propagation phase aligns with what other researchers have dubbed the “ca-tau-strophe,” which is when tangle pathology really begins to take off. “Our data suggest the ITG is the gateway to this cataustrophe,” he said. Seeley believes amyloid-targeted therapies will be most effective if they can prevent tau propagation from reaching this point of no return.
However, the researchers noted that among Aβ-positive people with MCI—a group eligible for aducanumab (Aduhelm) treatment—only 14 percent were in this earlier phase of tangle spread, while the rest were already in the propagation phase.
“These findings raise the possibility that a molecular-anatomical definition of disease stage may outperform clinical labels in predicting clinical responsiveness to amyloid-lowering and other AD therapies,” the authors wrote. Franzmeier and colleagues think stratifying patients based on their phase of tau spreading could be useful for clinical trials. They agree that removing Aβ before tau has spread may better prevent tau propagation than doing it afterward, and therefore is more likely to stave off neurodegeneration and cognitive decline.
Different Roads to Tauopathy
Much as the mind craves simplicity, these timed, anatomical stages of tau spreading may not apply to all people with AD. Scientists who study AD progression agree that Aβ accumulation instigates the spread of tangles from the entorhinal cortex into nearby regions in the mTL and eventually into the neocortex. However, according to Mormino’s study, there are exceptions.
First author Christina Young and colleagues studied regional patterns of tau deposition among people at the preclinical stage of AD, who had yet to exhibit overt cognitive impairment. The scientists tapped baseline amyloid- and tau-PET imaging data from the A4 study, a secondary prevention trial testing solanezumab in people who were cognitively normal at baseline. Of the 447 participants enrolled, 392 were amyloid-positive. Among them, 152 also had begun to accumulate neurofibrillary tangles and for most, these tangles were confined to the mTL. However, 36 volunteers deviated from this; they had tangles in cortical regions as well. These outliers could be subdivided into three groups based on their preponderance of accumulation: asymmetrical left, precuneus-dominant, and asymmetrical right. Importantly, these three patterns cropped up with similar prevalence in ADNI, the Harvard Aging Brain Study, and the Wisconsin Alzheimer’s Disease Research Center cohorts.
Although memory scores were similar between people with mTL versus cortical tau, those with cortical tau did worse on tests of executive function. Both people with mTL and those with cortical tangles had smaller hippocampi than controls. Among those with cortical deposits, the researchers spotted cortical thinning, though the thinning was not in regions with the most tangles. These amyloid-positive participants with cortical tangles tended to be younger than their counterparts with mTL tangles, suggesting that perhaps their disease was progressing faster.
In another mTL-sparing variation on this theme, Guo and colleagues reported that ADNI participants who were amyloid-positive by PET, but -negative by CSF, had significant tangle accumulation in outer cortical regions, but not in the mTL. In contrast, those who were amyloid-positive via CSF but not PET tended to have tau deposits only in the mTL. The findings imply that differences in Aβ accumulation may dictate where tau tangles accumulate in the preclinical stage of AD, and that mTL-tau measurements might miss the cortical tau tangles that accumulate among people with a “PET-first” Aβ pathway.
How do these different patterns of tau progression line up with those identified in the Korean/UCSF study? Young noted that not all participants with early Alzheimer’s disease in that study conformed to the author's proposed spreading framework. “These non-conforming participants may indeed be the ones with divergent cortical tau patterns highlighted in our study,” she wrote. “The same remote and local amyloid-tau interactions identified by Lee et al. may be occurring in those with divergent cortical tau, but the specific location of amyloid-tau interactions may vary outside of entorhinal cortex, leading to the spatial heterogeneity that we and others have described,” Young added (full comment below; Apr 2021 news). This could explain the known heterogeneity observed in various clinical presentations of Alzheimer’s disease (Ossenkoppele et al., 2016).
Franzmeier and colleagues made a similar point. “It will be crucial to test whether the proposed model of remote and local Aβ vs. tau interactions can also explain the heterogeneity in tau spreading, which may ultimately give rise to the clinically heterogeneous manifestation of AD, including ‘atypical’ AD phenotypes such as posterior cortical atrophy, dysexecutive, or language variant AD, all characterized by highly heterogeneous tau deposition patterns,” they wrote.
That such heterogenous tau patterns already emerge in asymptomatic people is new and interesting, they added. “This finding does have important implications for clinical trials, because individual variation in tau patterning may be indicative of individual variation in treatment response or disease progression, potentially in a cognitive-domain specific manner.”—Jessica Shugart
References
News Citations
- Tau PET Aligns Spread of Pathology with Alzheimer’s Staging
- Imaging Clinches Causal Connections between Aβ, Tau, Circuitry, and Cognition
- Forget Typical Alzheimer's: AI Finds Four Types.
Paper Citations
- Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife. 2019 Sep 2;8 PubMed.
- Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
Further Reading
Primary Papers
- Young CB, Winer JR, Younes K, Cody KA, Betthauser TJ, Johnson SC, Schultz A, Sperling RA, Greicius MD, Cobos I, Poston KL, Mormino EC, Alzheimer’s Disease Neuroimaging Initiative and the Harvard Aging Brain Study. Divergent Cortical Tau Positron Emission Tomography Patterns Among Patients With Preclinical Alzheimer Disease. JAMA Neurol. 2022 Jun 1;79(6):592-603. PubMed.
- Lee WJ, Brown JA, Kim HR, La Joie R, Cho H, Lyoo CH, Rabinovici GD, Seong JK, Seeley WW, Alzheimer’s Disease Neuroimaging Initiative. Regional Aβ-tau interactions promote onset and acceleration of Alzheimer's disease tau spreading. Neuron. 2022 Jun 15;110(12):1932-1943.e5. Epub 2022 Apr 19 PubMed.
- Jiang C, Wang Q, Xie S, Chen Z, Fu L, Peng Q, Liang Y, Guo H, Guo T, Alzheimer’s Disease Neuroimaging Initiative. β-Amyloid discordance of cerebrospinal fluid and positron emission tomography imaging shows distinct spatial tau patterns. Brain Commun. 2022;4(2):fcac084. Epub 2022 Mar 31 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Pennsylvania
VU University Medical Center
Ludwig Maximilians University
Lund University
This timely and well-executed study by Christina Young and colleagues focuses on divergence from the common regional expression of Alzheimer’s disease tau pathology during the preclinical disease stages.
Much about the AD pathological cascade appears to follow a fairly stereotyped progression, but there are many exceptions. The typical progression of AD tau has been famously codified into the Braak staging system (Braak et al., 2006), but seminal autopsy work has drawn attention to the fact that many individuals express regional tau patterns that do not fit neatly into this staging regime (Murray et al., 2011).
Meanwhile, AD patients presenting at the clinic with primary or predominating non-amnestic syndromes that defy the typical AD presentation are not uncommon, particularly in early onset cases where about a third of patients have non-amnestic AD. Tau-PET studies have begun to bridge these findings, showing that atypical, e.g., visual- or language-predominant clinical presentations of AD, often show unique regional patterns of tau pathology (Ossenkoppele et al., 2016).
Recent large tau-PET studies by our group have further supported the notion that individual differences abound in the expression of tau pathology (Franzmeier et al., 2020), and that these differences may manifest as systematic AD tau subtypes (Vogel et al., 2021). Studying this variation may be important for individualized treatment and may reveal biological insights about AD pathogenesis, but it is also important for the development of generalizable biomarkers that capture AD tau pathology across all of its regional manifestations.
Like some of the aforementioned studies, Young and co-authors explore individual heterogeneity in tau-PET patterns within an aggregated multi-cohort sample that features very little overlap with previous studies. However, the present study uniquely focuses exclusively on clinically unimpaired old adults, resembling the population likely to be recruited in the next generation of AD drug trials that aim to intervene before symptom onset. That heterogeneous tau patterns emerge in this asymptomatic population is novel and interesting, and demonstrates that deviations from the stereotypical Braak staging system already occur during the earliest stages of tau accumulation. This finding does have important implications for aforementioned clinical trials, as individual variation in tau patterning may be indicative of similar variation in treatment response or disease progression (potentially in a cognitive-domain specific manner).
This excellent study concludes that the frequency of atypical tau patterns is around 9 percent, which the authors say coincides well with one estimate of prevalence of atypical late-onset AD (6 percent). It is worth noting that this estimate was among all amyloid-positive individuals—the prevalence of atypical tau patterns was 23.6 percent among amyloid- and tau-positive individuals.
It is important to acknowledge that these estimates can depend highly on the definition of the entities in question. For example, the present study quantifies “abnormal” as 3 standard deviations from an a priori control group, whereas our previous work used a threshold of 2 SDs from a data-driven control group and, unsurprisingly, found a higher proportion of individuals with deviant tau patterns. Similarly, there is a great deal of heterogeneity in cognitive deficits among MCI and AD patients (Scheltens et al., 2017; Groot et al., 2021) that may be considered “atypical” depending on the definition. However, the authors make an excellent point that our field’s collective focus on amnestic presentations, particularly in MCI, may bias our estimates, and even our definitions of what is “typical.”
One other interesting finding among a great many in this study was that MRI indices of neurodegeneration were not consistently informative toward tau patterns. Hippocampal volume was reduced in both “typical” and “atypical” tau groups, and reductions in cortical thickness did not match regions of tau accumulation. This may be yet another source of individual variation (Das et al., 2021), perhaps due to premorbid differences in brain structure and the influence of co-pathologies. These findings suggest that MRI alone may not be sufficient to serve as a proxy of tau subtypes in asymptomatic individuals.
There is still a great deal to learn about heterogeneity in the progression of tau pathology, and what promise it may hold for improving our understanding of AD. Longitudinal studies will help this pursuit, as will better standardization of what is considered abnormal, and a continuation of trends toward collaboration across different research groups. This excellent study from Dr. Mormino’s group represents an important and well-presented step along this path!
References:
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006 Oct;112(4):389-404. PubMed.
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011 Sep;10(9):785-96. PubMed.
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
Franzmeier N, Dewenter A, Frontzkowski L, Dichgans M, Rubinski A, Neitzel J, Smith R, Strandberg O, Ossenkoppele R, Buerger K, Duering M, Hansson O, Ewers M. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer's disease. Sci Adv. 2020 Nov;6(48) Print 2020 Nov PubMed.
Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, Alzheimer’s Disease Neuroimaging Initiative, Pontecorvo MJ, Devous MD, Rabinovici GD, Alexander DC, Lyoo CH, Evans AC, Hansson O. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021 May;27(5):871-881. Epub 2021 Apr 29 PubMed.
Scheltens NM, Tijms BM, Koene T, Barkhof F, Teunissen CE, Wolfsgruber S, Wagner M, Kornhuber J, Peters O, Cohn-Sheehy BI, Rabinovici GD, Miller BL, Kramer JH, Scheltens P, van der Flier WM, Alzheimer's Disease Neuroimaging Initiative, German Dementia Competence Network, University of California San Francisco Memory and Aging Center, Amsterdam Dementia Cohort. Cognitive subtypes of probable Alzheimer's disease robustly identified in four cohorts. Alzheimers Dement. 2017 Apr 17; PubMed.
Groot C, Grothe MJ, Mukherjee S, Jelistratova I, Jansen I, van Loenhoud AC, Risacher SL, Saykin AJ, Mac Donald CL, Mez J, Trittschuh EH, Gryglewski G, Lanzenberger R, Pijnenburg YA, Barkhof F, Scheltens P, van der Flier WM, Crane PK, Ossenkoppele R. Differential patterns of gray matter volumes and associated gene expression profiles in cognitively-defined Alzheimer's disease subgroups. Neuroimage Clin. 2021;30:102660. Epub 2021 Apr 3 PubMed.
Das SR, Lyu X, Duong MT, Xie L, McCollum L, de Flores R, DiCalogero M, Irwin DJ, Dickerson BC, Nasrallah IM, Yushkevich PA, Wolk DA, Alzheimer's Disease Neuroimaging Initiative. Tau-Atrophy Variability Reveals Phenotypic Heterogeneity in Alzheimer's Disease. Ann Neurol. 2021 Nov;90(5):751-762. Epub 2021 Oct 15 PubMed.
View all comments by Oskar HanssonLudwig Maximilians University
University of Pennsylvania
VU University Medical Center
Lund University
Regional Aβ-tau interactions promote onset and acceleration of Alzheimer’s disease tau spreading
Alzheimer’s disease is characterized by cortical Aβ plaque pathology, followed by the successive spread of tau pathology throughout the brain, ensuing neurodegeneration and cognitive decline. Due to this cascade of pathological brain changes, AD is often considered an Aβ-induced tauopathy. Yet, the initial deposition patterns of Aβ and tau show a remarkable spatial mismatch (Jagust, 2018), which has posed challenges for developing a plausible mechanistic model of how Aβ may induce tau spreading in AD. In this elegant multimodal neuroimaging study jointly led by Bill Seeley and Joon-Kyung Seong, first author Wha Jin Lee proposes an integrative connectivity-based model that links cortical Aβ deposition with the successive spread of tau pathology throughout the brain.
The authors suggest two critical events that give rise to the cortical spreading of tau pathology in AD: As a “first hit,” cortical Aβ may induce tau spreading from the entorhinal cortex to other temporal lobe regions via remote connections. As a “second hit,” the local convergence of Aβ and tau in the inferior temporal lobe may induce widespread and accelerated tau spreading to the rest of the brain which may then drive the development of neurodegeneration and cognitive decline in AD (La Joie et al., 2020; Biel et al., 2021).
Together, the study is of high interest for the field, as the authors propose an integrated framework for how Aβ and tau pathology may interact i) remotely via neuronal connections as well as ii) locally to give rise to the brain-wide spreading pattern of tau pathology that is seen in AD.
Yet, these findings also raise several further issues to be addressed in the future: First, it will be critical to determine the underlying molecular and cellular mechanisms by which cortical Aβ induces remote tau spreading from the entorhinal cortex. Some proposed hypotheses include changing neuronal activity levels in the target region (Busche et al., 2008), which may facilitate trans-synaptic tau propagation (Wu et al., 2016), or by changing biophysical properties of tau.
These questions are difficult to address with neuroimaging methods alone, hence reverse translational studies using preclinical models will be necessary. Here, it will also be important to assess what differentiates local from remote Aβ and tau interactions, i.e., why local Aβ and tau interactions may potentiate and accelerate the subsequent spreading of tau pathology while remote Aβ vs. tau interactions have relatively circumscribed effects on tau spreading. It will be essential to understand the mechanisms that link Aβ and tau in order to identify potential novel treatment targets to “uncouple” Aβ deposition from tau spreading as early as possible in the course of AD.
Second, it remains to be clarified how the proposed Aβ-centric tau spreading model aligns with the earliest expansion of tau along medial temporal lobe (MTL) subregions (Berron et al., 2021). Earliest tau is typically seen in the entorhinal cortex and subsequently in the hippocampus and other MTL regions (Berron et al., 2021). Yet, these MTL regions typically don’t harbor extensive Aβ plaque pathology, so it is unlikely that MTL Aβ attracts tau from the connected entorhinal cortex. Of note, tau deposition in the MTL is also observed in primary age-related tauopathy (PART) in the absence of Aβ (Crary et al., 2014), thus MTL tau spreading may be driven by an Aβ-independent mechanism that is not captured by the current model.
Third, recent studies have emphasized considerable spatial heterogeneity in tau deposition and spreading in AD (Vogel et al., 2021; Franzmeier et al., 2020; Ossenkoppele et al., 2016), which does not adhere to the stereotypical “Braak-like” tau deposition pattern (Braak and Braak, 1991). Thus, it will be crucial to test whether the proposed model of remote and local Ab vs. tau interactions can also explain the heterogeneity in tau spreading which may ultimately give rise to the clinically heterogeneous manifestation of AD, including “atypical” AD phenotypes such as posterior cortical atrophy, dysexecutive, or language variant AD, all characterized by highly heterogeneous tau deposition patterns (Vogel et al., 2021; Ossenkoppele et al., 2016; Graff-Radford et al., 2021). The UCSF cohort of atypical AD patients would have been a well-suited sample to test this question, hence we encourage the authors to validate their model in these patients in the future.
As a limitation, Aβ levels saturate rather early in AD patients, and almost the entire neocortex harbors significant Aβ plaque pathology once patients show a positive Aβ-PET scan, regardless of the clinical phenotype (La Joie et al., 2019; Jeon et al., 2019; Beaufils et al., 2014). The ubiquity of Aβ can make it challenging to reliably test local or remote interactions with tau pathology, hence it should be clarified whether heterogeneity in Aβ deposition patterns may contribute to the heterogeneity in tau spreading (Vogel et al., 2021; Franzmeier et al., 2020; Ossenkoppele et al., 2016). Postmortem data suggest that initial sites of Aβ pathology vary (Thal et al., 2002), and sub-threshold Aβ has been shown to influence the degree of PET-assessed tau deposition in aging (Leal et al., 2018). Thus, it will be important to determine whether heterogeneous spatial deposition patterns of earliest “sub-threshold” Aβ (Leal et al., 2018) \may give rise to the subsequent heterogeneity in tau spreading patterns.
Lastly, we would like to highlight the studies’ approach to stage patients according to their respective “phase” of tau spreading, which allows identifying individuals in pre-acceleration or acceleration phases. We believe that this staging system can be relevant for participant stratification in clinical trials, since removing Aβ in the tau pre-acceleration phase may be more efficient in preventing downstream tau spreading and therefore neurodegeneration and cognitive decline.
Together, the current study is a major step forward in understanding the interaction between Aβ and tau pathology, which emphasizes the critical role of brain connectivity in the development of AD. The study also emphasizes the key role multi-modal neuroimaging can play to test and integrate mechanistic models of AD progression to better understand disease mechanisms. We believe this study will be an important starting point for future investigations to further disentangle the link between Aβ and tau pathology in AD by embedding both pathologies in a systems-level context of brain networks.
References:
Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 2018 Nov;19(11):687-700. PubMed.
La Joie R, Visani AV, Baker SL, Brown JA, Bourakova V, Cha J, Chaudhary K, Edwards L, Iaccarino L, Janabi M, Lesman-Segev OH, Miller ZA, Perry DC, O'Neil JP, Pham J, Rojas JC, Rosen HJ, Seeley WW, Tsai RM, Miller BL, Jagust WJ, Rabinovici GD. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020 Jan 1;12(524) PubMed.
Biel D, Brendel M, Rubinski A, Buerger K, Janowitz D, Dichgans M, Franzmeier N, Alzheimer’s Disease Neuroimaging Initiative (ADNI). Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res Ther. 2021 Aug 12;13(1):137. PubMed.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016 Aug;19(8):1085-92. Epub 2016 Jun 20 PubMed.
Berron D, Vogel JW, Insel PS, Pereira JB, Xie L, Wisse LE, Yushkevich PA, Palmqvist S, Mattsson-Carlgren N, Stomrud E, Smith R, Strandberg O, Hansson O. Early stages of tau pathology and its associations with functional connectivity, atrophy and memory. Brain. 2021 Mar 16; PubMed.
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014 Dec;128(6):755-66. Epub 2014 Oct 28 PubMed.
Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, Alzheimer’s Disease Neuroimaging Initiative, Pontecorvo MJ, Devous MD, Rabinovici GD, Alexander DC, Lyoo CH, Evans AC, Hansson O. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021 May;27(5):871-881. Epub 2021 Apr 29 PubMed.
Franzmeier N, Dewenter A, Frontzkowski L, Dichgans M, Rubinski A, Neitzel J, Smith R, Strandberg O, Ossenkoppele R, Buerger K, Duering M, Hansson O, Ewers M. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer's disease. Sci Adv. 2020 Nov;6(48) Print 2020 Nov PubMed.
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
Graff-Radford J, Yong KX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM, Jones DT, Murray ME. New insights into atypical Alzheimer's disease in the era of biomarkers. Lancet Neurol. 2021 Mar;20(3):222-234. PubMed.
La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, Doré V, Grinberg LT, Huang E, Hwang JH, Ikonomovic MD, Jack C Jr, Jagust WJ, Jin LW, Klunk WE, Kofler J, Lesman-Segev OH, Lockhart SN, Lowe VJ, Masters CL, Mathis CA, McLean CL, Miller BL, Mungas D, O'Neil JP, Olichney JM, Parisi JE, Petersen RC, Rosen HJ, Rowe CC, Spina S, Vemuri P, Villemagne VL, Murray ME, Rabinovici GD. Multisite study of the relationships between antemortem [11C]PIB-PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement. 2019 Feb;15(2):205-216. Epub 2018 Oct 19 PubMed.
Jeon S, Kang JM, Seo S, Jeong HJ, Funck T, Lee SY, Park KH, Lee YB, Yeon BK, Ido T, Okamura N, Evans AC, Na DL, Noh Y. Topographical Heterogeneity of Alzheimer's Disease Based on MR Imaging, Tau PET, and Amyloid PET. Front Aging Neurosci. 2019;11:211. Epub 2019 Aug 20 PubMed.
Beaufils E, Ribeiro MJ, Vierron E, Vercouillie J, Dufour-Rainfray D, Cottier JP, Camus V, Mondon K, Guilloteau D, Hommet C. The Pattern of Brain Amyloid Load in Posterior Cortical Atrophy Using (18)F-AV45: Is Amyloid the Principal Actor in the Disease?. Dement Geriatr Cogn Dis Extra. 2014 Sep;4(3):431-41. Epub 2014 Nov 11 PubMed.
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002 Jun 25;58(12):1791-800. PubMed.
Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ. Subthreshold Amyloid Predicts Tau Deposition in Aging. J Neurosci. 2018 May 9;38(19):4482-4489. Epub 2018 Apr 23 PubMed.
View all comments by Oskar HanssonStanford University School of Medicine
Both our paper and the Lee et al. paper tackle complementary questions relating to tau spread. While our study demonstrates that about 9 percent of clinically unimpaired individuals with abnormal Aβ have divergent cortical tau patterns, Lee et al. investigated the important question of how amyloid triggers tau spread when amyloid and tau follow different spatial patterns of spread, i.e., amyloid begins in the neocortex and spreads into the entorhinal cortex in Phase 2 (Thal et al., 2002), while tau begins in the transentorhinal region and spreads to entorhinal cortex and eventually neocortex (Braak et al., 2006).
Lee et al. provide evidence for both remote and local amyloid-tau interactions, such that first there are remote interactions between cortical amyloid and entorhinal tau that promote tangle spread to nearby regions connected to entorhinal cortex, then tau deposits reach the inferior temporal gyrus where they locally interact with amyloid for the first time. Finally, tangles spread into amyloid-positive neocortical regions that are connected to the inferior temporal gyrus.
That Lee et al. identify the inferior temporal gyrus as a key region for amyloid-tau interactions is intriguing, given that our study also suggested that inferior temporal, lateral parietal, and precuneus may be especially vulnerable areas for early tau deposition. Indeed, consistent with our suggestion, Figure 4B in the Lee et al. paper shows that lateral parietal and precuneus, in addition to inferior temporal gyrus, also show high local and remote amyloid and tau interactions. Together, our studies highlight the importance of these regions in models of Alzheimer’s disease progression.
Lee et al. also noted that not all participants with early Alzheimer’s disease conformed to their spreading framework, highlighting the existence of heterogeneity across individuals. These non-conforming participants may indeed be the ones with divergent cortical tau patterns highlighted in our study. The same remote and local amyloid-tau interactions identified by Lee et al. may be occurring in those with divergent cortical tau, but the specific location of amyloid-tau interactions may vary outside of the entorhinal cortex, leading to the spatial heterogeneity that we and others (Vogel et al., 2021) have described.
This model would provide an explanation for the known heterogeneity observed in various clinical presentations of Alzheimer’s disease (Ossenkoppele et al., 2016). In other words, the mechanisms related to local and remote amyloid-tau interactions could be similar across clinical presentations of Alzheimer’s disease, but individual differences in cortical hub regions could result in heterogeneous patterns of tau spread, which could in turn give rise to different clinical symptom profiles. The reasons why this heterogeneity occurs beyond entorhinal cortex remains unknown and is a key unanswered gap in Alzheimer’s disease research.
References:
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002 Jun 25;58(12):1791-800. PubMed.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006 Oct;112(4):389-404. PubMed.
Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, Alzheimer’s Disease Neuroimaging Initiative, Pontecorvo MJ, Devous MD, Rabinovici GD, Alexander DC, Lyoo CH, Evans AC, Hansson O. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021 May;27(5):871-881. Epub 2021 Apr 29 PubMed.
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
Mayo Clinic
These interesting papers deal with important questions regarding the spatial and temporal discrepancies between amyloid and tau that appeal to mechanisms that relate to the functional architecture of the brain. The authors are also trying to integrate the well-known phenomenon of phenotypic heterogeneity in clinical presentations, structural imaging, molecular imaging, functional imaging, and pathology (Graff-Radford et al., 2021).
These are topics we have previously synthesized within the cascading network failure model of Alzheimer’s disease and subsequently investigated in aging and across AD phenotypes (Jones et al., 2017; Jones et al., 2016; Sintini et al., 2021; Wiepert et al., 2017). In our studies, we use functional connectivity measures from patients rather than using templates from healthy controls, which allows for a different view of the functional physiology and its relationship to amyloid and tau reported in most studies. Our model emphasizes distributed functional physiology in ensembles of cells spanning large-scale anatomy associated with mental functions, or the global functional state space (GFSS) (Jones et al., 2022). In our study of the GFSS, we observed that there is a relatively simple relationship between mental functions and brain anatomy that can predict many aspects of Alzheimer’s physiology, including Braak staging of tau neurofibrillary tangle pathology. This underlying functional organization may drive many of the relationships reported in associative neuroimaging studies using large-scale anatomic patterns across all degenerative disease that cause dementia. That is why we were also able to relate this simple brain-behavior mapping to large-scale brain networks, mental task activation patterns, and a diverse array of clinical syndromes that span the dementia spectrum. In our model, large scale neurodynamics that take place across the landscape of the GFSS is the key element that links functional physiology to selective patterns of degenerative anatomy. In this model, neurodegenerative selectivity for certain dynamic brain patterns, or modes of function of the complex information processing system in the brain, requires a fundamental role for large-scale neurodynamic physiology in AD and related disorders.
These functional dynamics that influence cellular activity across large ensembles of cells may contribute to homeostatic failure in protein processing physiology. This is consistent with an accelerated failure time model of amyloid and tau (Therneau et al., 2021). This alternative view of the relationship between amyloid, tau, and functional brain organization suggests different biomarkers and therapeutic targets related to large-scale functional physiology that can complement existing models focused more on molecular behavior.
References:
Graff-Radford J, Yong KX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM, Jones DT, Murray ME. New insights into atypical Alzheimer's disease in the era of biomarkers. Lancet Neurol. 2021 Mar;20(3):222-234. PubMed.
Jones DT, Graff-Radford J, Lowe VJ, Wiste HJ, Gunter JL, Senjem ML, Botha H, Kantarci K, Boeve BF, Knopman DS, Petersen RC, Jack CR Jr. Tau, amyloid, and cascading network failure across the Alzheimer's disease spectrum. Cortex. 2017 Dec;97:143-159. Epub 2017 Oct 3 PubMed.
Jones DT, Knopman DS, Gunter JL, Graff-Radford J, Vemuri P, Boeve BF, Petersen RC, Weiner MW, Jack CR Jr, Alzheimer’s Disease Neuroimaging Initiative. Cascading network failure across the Alzheimer's disease spectrum. Brain. 2016 Feb;139(Pt 2):547-62. Epub 2015 Nov 19 PubMed.
Sintini I, Graff-Radford J, Jones DT, Botha H, Martin PR, Machulda MM, Schwarz CG, Senjem ML, Gunter JL, Jack CR, Lowe VJ, Josephs KA, Whitwell JL. Tau and Amyloid Relationships with Resting-state Functional Connectivity in Atypical Alzheimer's Disease. Cereb Cortex. 2021 Feb 5;31(3):1693-1706. PubMed.
Wiepert DA, Lowe VJ, Knopman DS, Boeve BF, Graff-Radford J, Petersen RC, Jack CR Jr, Jones DT. A robust biomarker of large-scale network failure in Alzheimer's disease. Alzheimers Dement (Amst). 2017;6:152-161. Epub 2017 Jan 25 PubMed.
Jones D, Lowe V, Graff-Radford J, Botha H, Barnard L, Wiepert D, Murphy MC, Murray M, Senjem M, Gunter J, Wiste H, Boeve B, Knopman D, Petersen R, Jack C. A computational model of neurodegeneration in Alzheimer's disease. Nat Commun. 2022 Mar 28;13(1):1643. PubMed.
Therneau TM, Knopman DS, Lowe VJ, Botha H, Graff-Radford J, Jones DT, Vemuri P, Mielke MM, Schwarz CG, Senjem ML, Gunter JL, Petersen RC, Jack CR Jr. Relationships between β-amyloid and tau in an elderly population: An accelerated failure time model. Neuroimage. 2021 Nov 15;242:118440. Epub 2021 Jul 29 PubMed.
Make a Comment
To make a comment you must login or register.