Induced Neurons From AD Patients Hint at Disease Mechanisms
Quick Links
Human neurons, whether made from induced pluripotent stem cells (iPSCs) or directly from fibroblasts, are one of the newest tools for Alzheimer’s disease research. They hold the potential to model human disease and provide clues to the etiology of sporadic AD, but they are just now taking their first baby steps in that direction. In a Nature paper published online January 25, researchers led by Lawrence Goldstein at the University of California, San Diego, follow last year’s reports of neurons made from presenilin mutation carriers with the first published report of neurons generated from people with APP mutations and from those with sporadic AD. Though these cells come from only a handful of patients so far, they offer tantalizing hints to human pathology and strengthen previous links between AD pathology and protein cycling. Interestingly, the authors also turn up evidence that tau pathology occurs in response to amyloid precursor protein (APP) processing by β-secretase (BACE1), but not by γ-secretase. This suggests that Aβ and tau pathology may arise from separate APP cleavage fragments. If this finding holds up, it would have implications for clinical treatments.
The first report of neurons made from familial AD patients came out last year, from Asa Abeliovich’s group at Columbia University, New York City (see ARF related news story on Qiang et al., 2011). More recently, a Japanese group has also described the generation of human AD neurons (see Yagi et al., 2011). Both these studies used cells from people with familial presenilin mutations.
In contrast, Goldstein and colleagues took cells from two familial AD patients who had an APP duplication, two patients with sporadic disease, and two healthy, age-matched controls. None of the patients carried the allele for ApoE4, the primary genetic risk factor for sporadic AD. First author Mason Israel generated three iPSC lines from each patient’s fibroblasts, then differentiated the cells and separated them by fluorescence-activated cell sorting to achieve pure neuronal populations. These cultures contained a mix of glutamatergic and GABAergic cells; a future goal is to achieve pure populations of specific neuronal types, Goldstein told ARF.
Compared to neurons made from age-matched donors, neurons made from both of the familial AD patients, as well as from one of the sporadic patients, showed an AD-like phenotype, secreting large amounts of Aβ40 and stockpiling phosphorylated tau, as measured in cell lysates. Aβ42 levels were below detection limits in these cultures, due to the low number of cells, the authors report. This contrasts with Abeliovich’s findings of an increased Aβ42/Aβ40 ratio in induced neurons derived from patients with presenilin mutations. The APP mutant neurons and the same sporadic AD line also produced high levels of activated GSK-3β, a kinase that phosphorylates tau, compared to controls.
Intriguingly, these three neuron lines developed large endosomes, a phenomenon seen also in Abeliovich’s study and in autopsy samples from patients with AD and Down’s syndrome (see, e.g., Nixon, 2005). “The interesting possibility is that altered endocytosis is an early phenotype that leads to abnormal processing and downstream events,” Goldstein remarked. Several studies have turned up genetic links between trafficking and endocytic proteins, such as SORLA and SorCS1, and AD (see, e.g., ARF related news story, ARF news story, and ARF news story).
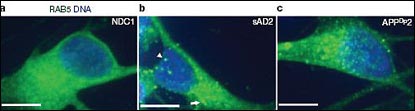
Induced AD neurons: Compared to control neurons (left image), induced neurons made from people with sporadic AD (middle) and familial AD (right) accumulate large endosomes as seen by synapsin 1 labeling (green). Image courtesy Lawrence Goldstein, UCSD, and Nature
The authors wondered whether altered APP processing would affect tau, as a number of papers report that tau tangles occur downstream of Aβ pathology (see, e.g., ARF related news story on Roberson et al., 2007). Since successive cleavage of APP by BACE1 and γ-secretase produces Aβ, the researchers treated the neuronal cultures from both sporadic and familial patients with either BACE1 inhibitors or γ-secretase inhibitors for 24 hours, and then measured levels of phosphorylated tau and activated GSK-3β. Although inhibiting γ-secretase had no effect, inhibiting BACE1 lowered levels of both phosphorylated tau and activated GSK-3β. The results imply that a BACE cleavage product other than Aβ may affect downstream tau pathology, the authors note. One candidate molecule is the C-terminal fragment produced by BACE cleavage, β-CTF, which has been implicated in endosome accumulation and in axonopathies (see ARF related news story and ARF news story on Salehi et al., 2006). Goldstein told ARF he plans to follow up on this result with mechanistic studies to try to pinpoint exactly what fragment is affecting tau.
Scott Small at Columbia pointed out that this finding lends support to the hypothesis that, rather than tau being directly downstream of Aβ, a common upstream element drives both pathologies through separate pathways (see Small and Duff, 2008). “The clinical implications of this are, of course, important, as it would suggest that Aβ-lowering drugs might not be sufficient to ameliorate tau pathology,” Small wrote to ARF.
Abeliovich called the paper exciting, and noted it is the first study to use AD neurons derived from iPS cells to analyze tau-related changes. “Given the differences in tau between rodents and humans, it makes the use of human iPS cells especially relevant,” he said. One limitation of iPSC technology, however, is that the large genetic variation between individuals makes it difficult to be sure which genes are causing an observed phenotype. The next step in the APP mutant lines would be to knock down APP and see if the levels of Aβ40 and phosphorylated tau drop, Abeliovich suggested. He also noted that this study is based on very few cell lines, so the data are preliminary. Goldstein said his goal is to scale up the technology so that he can look at cell lines from 50 or 100 patients instead of six.
Goldstein is intrigued by the fact that neurons made from one of the patients with sporadic AD behaved like the neurons from familial patients. One possibility is that this patient carried unknown genetic risk factors for AD. The results need to be repeated, Goldstein noted. In particular, he would like to find out what percentage of people with sporadic AD produce neurons that misbehave in a dish. If the percentage is significant, it would hint that iPS cells might one day be used as a prospective diagnostic. “Maybe this is a window to evaluate individual risk,” Goldstein speculated. He is also interested in doing drug discovery in neurons derived from iPS cells, and in looking at contributions from other cell types such as astrocytes. “You can learn unexpected things from using [iPSC] technology and working on humans,” he said. “I think it’s a transformative technology for understanding both basic biology and how it links to the development of disease.”—Madolyn Bowman Rogers
References
News Citations
- Alzheimer’s Neurons Made to Order: Direct Conversion From Skin Cells
- Sorting Out SorLA—What Role in APP Processing, AD?
- Aβ and SORLA—Partners in Cerebrovascular Crime?
- APP Sorting Protein May Link Alzheimer’s and Diabetes
- APP Mice: Losing Tau Solves Their Memory Problems
- APP in Pieces: βCTF implicated in Endosome Dysfunction
- Trisomy Trouble: Neurotrophin Signaling Defective in Down Syndrome
Paper Citations
- Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, Moreno H, Abeliovich A. Directed conversion of Alzheimer's disease patient skin fibroblasts into functional neurons. Cell. 2011 Aug 5;146(3):359-71. PubMed. RETRACTED
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum Mol Genet. 2011 Dec 1;20(23):4530-9. PubMed.
- Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005 Mar;26(3):373-82. PubMed.
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed.
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006 Jul 6;51(1):29-42. PubMed.
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008 Nov 26;60(4):534-42. PubMed.
Further Reading
News
- Alzheimer’s Neurons Made to Order: Direct Conversion From Skin Cells
- Sorting Out SorLA—What Role in APP Processing, AD?
- ApoE(ε)4 Brains Have to Work Harder
- APP Sorting Protein May Link Alzheimer’s and Diabetes
- APP Mice: Losing Tau Solves Their Memory Problems
- APP in Pieces: βCTF implicated in Endosome Dysfunction
- Trisomy Trouble: Neurotrophin Signaling Defective in Down Syndrome
Primary Papers
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012 Jan 25;482(7384):216-20. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Lund University
The use of human induced pluripotent stem (iPS) and induced neuronal (iN) cells is clearly a major step forward compared to standard clonal cell lines, and also to our own mainstay system, cultured primary AD transgenic neurons. However, the results with induced cells are not yet all that easy to fully interpret. Findings of an elevated Aβ42/40 ratio (Qiang et al., 2011; Yagi et al., 2011) fit well with what is known from prior work on familial AD (FAD) mutations. The enlarged early endosomes seen both by Qiang et al., and now Israel et al., also fit well with pioneering work by Ralph Nixon and Ann Cataldo. Yet it is still unclear if these genetically engineered cells are equivalent to primary human neurons. Israel and colleagues’ evidence that tau is altered by C-terminal fragments (CTFs) of amyloid precursor protein (APP) rather than Aβ builds on a growing literature of Aβ-independent effects due to FAD mutations (here, APP duplications). However, these observations are based on treatment with γ-secretase inhibitors. which can be tricky because of potential effects on iPS cell differentiation, toxicity, etc. In addition, a considerable number of divergent data point to Aβ rather than CTFs as being critical.
In our experience, limited (overnight) γ-secretase inhibition did prevent both alterations in synapses (Almeida et al., 2005; Tampellini et al., 2009) and endocytosis (Almeida et al., 2006), albeit in mutant APP-overexpressing transgenic neurons. In contrast, longer treatments (or higher concentrations) with γ-secretase inhibitor were not protective. Based on work in primary transgenic neurons, synapsin 1 would also not be the optimal marker to use for identifying presynaptic changes, which Israel et al. found to be unchanged in their AD iPS cells. It will be important to obtain more data with these exciting new model systems as noted by Abeliovich in the ARF news story.
References:
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, Moreno H, Abeliovich A. Directed conversion of Alzheimer's disease patient skin fibroblasts into functional neurons. Cell. 2011 Aug 5;146(3):359-71. PubMed. RETRACTED
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum Mol Genet. 2011 Dec 1;20(23):4530-9. PubMed.
Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005 Nov;20(2):187-98. PubMed.
Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009 Aug 5;29(31):9704-13. PubMed.
Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. PubMed.
Make a Comment
To make a comment you must login or register.