Does Soluble TREM2 Rile Up Microglia?
Quick Links
Microglial cells may ensure their own survival by cutting a receptor loose from their surface. As described February 16 in the Journal of Experimental Medicine, the soluble portion of triggering receptor expressed on myeloid cells 2, aka sTREM2, activates microglia and shields them from death. Researchers led by Guojun Bu at the Mayo Clinic in Jacksonville, Florida, also reported that mutations known to increase risk of Alzheimer’s disease sapped sTREM2’s protective and inflammatory prowess. The findings suggest that the freewheeling fragment of TREM2 counteracts the previously reported anti-inflammatory role of its membrane-bound parent.
While the role of sTREM2 in AD pathogenesis is unclear, Bu said he views its pro-inflammatory function as a double-edged sword. On one hand, sTREM2 may trigger microglia to remove Aβ. On the other, it may push the cells into chowing down on precious synapses, hastening neurodegeneration.
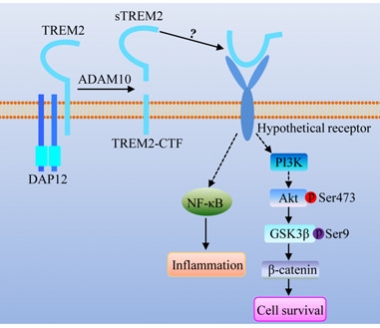
Soluble Siren.
Following its liberation from the cell surface, soluble TREM2 signals through an unknown receptor. [Image courtesy of Guojun Bu and Li Zhong.]
An immune receptor expressed on microglia, TREM2 is poised to steer responses of the brain’s primary immune cells. While the TREM2 field has been marked by complex and sometimes contradictory findings since researchers first linked it to AD risk (see Research Timeline 2012), the predominant view is that signaling through the receptor exerts an anti-inflammatory, pro-phagocytic effect. Various TREM2 ligands—including phospholipids and even ApoE—promote microglial survival and clearance of debris, including Aβ plaques (see Feb 2015 news; Jul 2016 conference news; Oct 2015 news). Thickening the plot, ADAM proteases can liberate the TREM2 ectodomain from the microglial surface. In cerebrospinal fluid, the concentration of this shed fragment rises in the prodromal phase of AD as well as in other neurodegenerative diseases, and tracks with the elevation of tau and p-tau (see Piccio et al., 2008; Apr 2015 conference news; Jan 2016 news). What part this soluble fragment plays in immune responses mediated by TREM2 in the healthy or diseased brain remains to be addressed.
To investigate, co-first authors Li Zhong and Xiao-Fen Chen of Xiamen University in China and colleagues started by generating a recombinant version of the TREM2 extracellular domain, fused to an antibody Fc fragment. This allowed them to purify sufficient quantities of sTREM2 for injection into animals or cell cultures. They then incubated primary mouse microglia with 20 nM of the fusion protein, and found that microglia survived just as well as they did when cultured with GM-CSF, an essential support factor for microglia in culture. Strikingly, sTREM2-Fc delivered the same survival benefit to TREM2 knock-out microglia, or to microglia lacking DAP12, the adaptor molecule through which full-length TREM2 signals. These findings indicated that sTREM2 did not signal via its own membrane-bound precursor. This is contrary to the prevailing idea that the soluble fragment functions primarily as a decoy receptor, hoarding available ligands away from its parent.
The researchers next explored how sTREM2 affects microglial activation. They found it ramped up the transcription of IL-1β, IL-6, TNF-α, and IL-10 in a dose-dependent manner. All these cytokines promote inflammatory responses. Conversely, sTREM2 did not budge expression of Arg-1 and Ym-1, two proteins known to dampen inflammation.
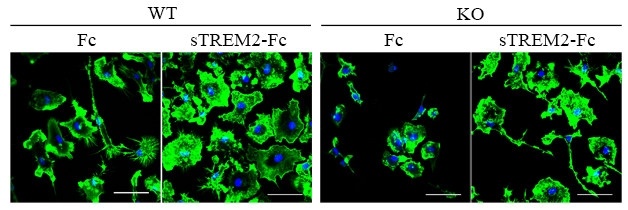
Morphing Microglia. Upon treatment with sTREM2-Fc, microglia from WT (left panels) or TREM2 KO mice (right panels) round up into a pro-inflammatory state. [Image courtesy of Guojun Bu and Li Zhong.]
Microglia treated with sTREM2-Fc also morphed into an activated form, pulling in the spindly processes they use to continually survey their environment and assuming a rounder shape. Just as it did for cell survival, sTREM2-Fc activated microglia that lacked TREM2 or DAP12, again hinting a separate signal transduction pathway was involved. The researchers observed similar effects on microglial survival and activation when they used an Fc-free version of human sTREM2, or when they used mouse sTREM2-Fc.
Microglia in the brain also responded to sTREM2. Compared to injection of Fc alone, injection of sTREM2-Fc into the hippocampi of normal or TREM2 knock-out mice stimulated the expression of pro-inflammatory cytokines, and coaxed microglia there to round up into their activated state.
Armed with a tool kit of inhibitors, the researchers homed in on the signaling pathways that might be responsible for sTREM2’s effects. They found that sTREM2 promoted microglial survival via the well-known anti-apoptotic Akt-GSK3β-β-catenin pathway, dovetailing with the lab’s recent report that knocking out TREM2 attenuates Akt/GSK3β signaling and microglial survival via this pathway (see Zheng et al., 2017). They also found that sTREM2 switched on pro-inflammatory cytokines via the transcription factor NF-κB.
How do AD risk variants fit into this picture? To find out, the researchers treated microglia with sTREM2-Fc fusion proteins harboring the R47H or R62H mutations in their extracellular domains. These mutations reportedly interfere with TREM2’s ability to bind various ligands. They found that both of these mutations lessened the protection and activation of microglia by sTREM2. Notably, these AD-linked mutations are distinct from two other missense mutations, T66M and Y38C, which are associated with frontotemporal dementia. Researchers led by Christian Haass of the German Center for Neurodegenerative Diseases in Munich had previously reported that these mutants bungled the transport of TREM2 to the cell surface and prevented shedding of sTREM2, while R47H had lesser effects on TREM2 maturation (see Kleinberger et al., 2014; Jul 2014 webinar).
Bu said his findings support the idea that the R47H and R62H variants elevate AD risk through a loss-of-function mechanism, perhaps thwarting sTREM2’s ability to enlist microglia in the clean-up of Aβ-laden debris. The pro-inflammatory effects of sTREM2 appear to run counter to the reported anti-inflammatory effects of full-length TREM2, supporting the idea that the soluble fragment is unleashed in response to injury, Bu said. He suggested that promoting sTREM2 function in the earliest stages of AD could be beneficial, although too much inflammation could turn harmful once neurodegeneration is initiated.
Monica Carson of the University of California, Riverside, interpreted the data slightly differently. She believes the soluble fragment may be crucial for mounting acute responses to injuries or infections that occur throughout life. “Maybe sTREM2 allows microglia to live longer in the face of various bouts of inflammation,” she said. However, in the face of chronic injury (such as Aβ aggregation), sTREM2 may do more harm than good. “Once chronic inflammation sets in, perhaps it would be better if those microglia didn’t survive,” she said. She suggested that blocking sTREM2 function, rather than promoting it, would have a better chance at slowing neurodegeneration.
“Although it is known that there is an increase in sTREM2 in many disease states, the role that sTREM2 plays is still relatively unknown,” wrote Marco Colonna of Washington University in St. Louis. “The data presented here provide a first step toward this goal, suggesting a protective function of sTREM2 on microglia in steady state.” He added that the study raises many questions about the nature of sTREM2’s role in disease, and what its receptor might be.
“Conceptually, these data support recent proposals that the presence of sTREM2, as detected in human biological samples, indicate microglial activation,” wrote Gary Landreth of Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, in a comment to Alzforum. “Unanswered is the question of whether sTREM2 has analogous deleterious effects on astrocytes and neurons, and what relationship sTREM2 has with full-length TREM2,” he added.—Jessica Shugart
References
News Citations
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- Unbiased Screen Fingers TREM2 Ligands That Promote Aβ Uptake
- Alzheimer’s Risk Genes Interact in Immune Cells
- Microglia—Who Are You Really? New Clues Emerge
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
Webinar Citations
Paper Citations
- Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J 2nd, Naismith RT, Panina-Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008 Nov;131(Pt 11):3081-91. Epub 2008 Sep 12 PubMed.
- Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD, Wang X, Zhang YW, Xu H, Bu G. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J Neurosci. 2017 Feb 15;37(7):1772-1784. Epub 2017 Jan 11 PubMed.
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Other Citations
Further Reading
Papers
- Wood H. Alzheimer disease: Soluble TREM2 in CSF sheds light on microglial activation in AD. Nat Rev Neurol. 2017 Feb;13(2):65. Epub 2017 Jan 13 PubMed.
Primary Papers
- Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L, Jia L, Zheng H, Painter M, Atagi Y, Liu CC, Zhang YW, Fryer JD, Xu H, Bu G. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017 Mar 6;214(3):597-607. Epub 2017 Feb 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Indiana University School of Medicine
Zhong et al. report that soluble forms of TREM2 are able to elicit a robust proinflammatory response and promote survival in primary microglia. These findings provide a completely novel take on the biological roles of soluble TREM2. Previous work has shown, using a similar sTREM2-Fc fusion protein, that soluble Trem2 disrupts Trem2 signaling involved in osteoclastogenesis (Kim et al., 2005). Since then, the field’s focus has shifted to the use of sTREM2 as a clinical biomarker for AD and other neurodegenerative diseases (Piccio et al., 2008; Piccio et al., 2016; Heslegrave et al., 2016; Suarez-Calvet et al., 2016). A homologous soluble factor, soluble Trem1 was demonstrated to reverse LPS-induced endotoxic shock by reducing proinflammatory cytokine levels and promoting survival of mice injected with murine Trem1-Fc fusion protein systemically (Bouchon et al., 2001). These findings, in light of the Zhong et al. paper, illustrate that soluble TREM proteins do modulate inflammation and survival mechanisms.
Zhong and colleagues show that sTREM2 acts in an autocrine manner to drive the expression of NFκB-dependent proinflammatory cytokine expression and microglial survival. These effects are independent of the endogenous microglial TREM2 receptor and its adapter DAP12. The central question arising from these findings is how the cleaved TREM2 extracellular domain interacts with primary microglia to elicit activation of the intracellular signaling cascades. Immunohistochemistry depicts a change in microglial phenotype characteristic of activation with both in vitro and in vivo application of sTREM2-Fc. Conceptually, these data support recent proposals that the presence of sTREM2, as detected in human biological samples, indicates microglial activation (Ohrfelt et al., 2016; Gispert et al., 2016a; Gispert et al., 2016; Suarez-Calvet et al., 2016; Heslegrave et al., 2016; Piccio et al., 2016). This new study begins to address the significance of sTREM2 release on microglial function and the overall innate immune response in the context of disease. A major caveat with previous studies employing sTREM2-Fc fusion protein to assess Trem2 function is that this chimeric protein may not mimic the physiological activity of Trem2 and its soluble form. This study is the first to validate their findings with sTREM2-Fc using an untagged form of sTREM2.
The primary data describe a very robust induction of IL-1β, IL-6 and TNFα mRNA levels within four hours following exposure to sTREM2. In the case of IL-6, mRNA levels rose more than 3,000 fold and IL-1β 120-fold, a response that is greater than that typically elicited by TLR4 stimulation and was highly variable between assays. However, protein levels were not quantitated and only normalized RNA values were provided.
One of the key findings of the study was the observation that disease-linked sTREM2 variants were about 50 percent less effective in suppressing apoptosis of microglia deprived of GM-CSF and of stimulating cytokine mRNA levels similar to WT sTREM2-Fc. Others have shown the particular variants tested, R47H and R62H, as either Trem2-Fc proteins (Yeh et al., 2016) or expressed within reporter cell lines (Song et al., 2016) to impede Trem2 recognition of apolipoproteins. Thus, these variants appear to impair ligand binding, which could explain the findings by Zhong et al. examining these variants when expressed in sTREM2-Fc constructs.
Unanswered is the question of whether sTREM2 has analogous deleterious effects on astrocytes and neurons, and what relationship sTREM2 has with full-length Trem2. Trem2 has historically been described as anti-inflammatory, but many studies now depict Trem2 as an amplifier of inflammatory responses under multiple contexts, including traumatic brain injury (Saber et al., 2017), ischemia (Sieber et al., 2013), and demyelination (Poliani et al., 2015). Some (Kleinberger et al., 2014; Piccio et al., 2008) have proposed sTREM2 to serve as a decoy receptor for full-length Trem2 that works antagonistically to Trem2, but this study shows for the first time that sTREM2 may complement and even amplify Trem2 signaling.
References:
Kim Y, Sato K, Asagiri M, Morita I, Soma K, Takayanagi H. Contribution of nuclear factor of activated T cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J Biol Chem. 2005 Sep 23;280(38):32905-13. Epub 2005 Jul 26 PubMed.
Piccio L, Deming Y, Del-Águila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016 Jun;131(6):925-33. Epub 2016 Jan 11 PubMed.
Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J 2nd, Naismith RT, Panina-Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008 Nov;131(Pt 11):3081-91. Epub 2008 Sep 12 PubMed.
Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Öhrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener. 2016 Jan 12;11:3. PubMed.
Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C, Dominantly Inherited Alzheimer Network. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016 Dec 14;8(369):369ra178. PubMed.
Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001 Apr 26;410(6832):1103-7. PubMed.
Öhrfelt A, Axelsson M, Malmeström C, Novakova L, Heslegrave A, Blennow K, Lycke J, Zetterberg H. Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult Scler. 2016 Jan 11; PubMed.
Gispert JD, Suárez-Calvet M, Monté GC, Tucholka A, Falcon C, Rojas S, Rami L, Sánchez-Valle R, Lladó A, Kleinberger G, Haass C, Molinuevo JL. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer's disease. Alzheimers Dement. 2016 Dec;12(12):1259-1272. Epub 2016 Jul 14 PubMed.
Gispert JD, Monté GC, Suárez-Calvet M, Falcon C, Tucholka A, Rojas S, Rami L, Sánchez-Valle R, Lladó A, Kleinberger G, Haass C, Molinuevo JL. The APOE ε4 genotype modulates CSF YKL-40 levels and their structural brain correlates in the continuum of Alzheimer's disease but not those of sTREM2. Alzheimers Dement (Amst). 2017;6:50-59. Epub 2016 Dec 22 PubMed.
Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C, Dominantly Inherited Alzheimer Network. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016 Dec 14;8(369):369ra178. PubMed.
Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Öhrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener. 2016 Jan 12;11:3. PubMed.
Piccio L, Deming Y, Del-Águila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016 Jun;131(6):925-33. Epub 2016 Jan 11 PubMed.
Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 2016 Jul 20;91(2):328-40. PubMed.
Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, Wang Y, Tanzi RE, Colonna M. Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017 Apr;13(4):381-387. Epub 2016 Aug 9 PubMed.
Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT. Triggering Receptor Expressed on Myeloid Cells 2 Deficiency Alters Acute Macrophage Distribution and Improves Recovery after Traumatic Brain Injury. J Neurotrauma. 2016 May 9; PubMed.
Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8(1):e52982. Epub 2013 Jan 3 PubMed.
Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015 May;125(5):2161-70. Epub 2015 Apr 20 PubMed.
Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J 2nd, Naismith RT, Panina-Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008 Nov;131(Pt 11):3081-91. Epub 2008 Sep 12 PubMed.
Washington University School of Medicine
The article represents a potentially interesting finding regarding the role of sTREM2 in triggering microglial activation. Although it is known that there is an increase in sTREM2 in many disease states, the role that sTREM2 plays is still relatively unknown. The data presented here provide a first step toward this goal, suggesting a protective function of sTREM2 on microglia in steady state. There are, however, a few outstanding questions that I have outlined below.
1. The authors do not identify a receptor, nor do they provide evidence regarding how sTREM2 may be exerting a protective effect.
2. The use of sTREM2-Fc is interesting and very valuable but is not overly physiological. Although the authors control for the Fc portion in many experiments it is unclear what effect tethering an Fc fragment may have on sTREM2. The authors do generate a TEV-Fc version of sTREM2, which can be cleaved resulting in a non-Fc version of sTREM2, but this “clean” sTREM2 was not used throughout the paper.
3. The in vivo experiment is a bit confusing. In WT mice there is likely to be some sTREM2 already available. It is difficult to understand why sTREM2-Fc has such a robust effect in WT mice and why sTREM2 generally elicits a very similar response in both WT and TREM2-/- mice. Again, these results would have been greatly strengthened if the authors had used their sTREM2 without an Fc.
4. Finally, the most important question, which is what role does sTREM2 play in disease, remains to be addressed. I guess the authors will follow this up soon.
Make a Comment
To make a comment you must login or register.