New Data Reinforces Concept of Protein Propagation
Quick Links
Research into frontotemporal dementias has powered forward in the last few years, propelled by a new understanding of the underlying biology of this group of diseases. Nonetheless, one major roadblock is the lack of good animal models. At the 10th International Conference on Frontotemporal Dementias, held August 31-September 2 in Munich, Virginia Lee of the University of Pennsylvania, Philadelphia, presented new data on an inducible model of TDP-43 pathology. It provided more clues to how the brain becomes less resilient to FTD with age, and generated excitement among attendees. Meanwhile, researchers at Ludwig-Maximilians University, Munich, debuted a new model of TREM2 deficiency. Other speakers focused on deciphering when disease starts and how it progresses, in particular on how pathologic proteins spread through the animal brain. Data from the UPenn group support the idea that misfolded proteins propagate, corrupting native protein along the way. Other scientists added human data on a vulnerable cell type in behavioral variant FTD, and highlighted different patterns of progression in bvFTD and amyotrophic lateral sclerosis (ALS). Researchers were hopeful that a deeper understanding of molecular mechanisms will one day lead to better treatments.
Building a Better Mouse Model
TDP-43 proteinopathy, seen in half of FTD and nearly all ALS cases, has been difficult to model in mice. Knockouts die because the protein plays an essential role in development, and overexpression models poorly reflect human disease (see Sep 2012 news). Lee and colleagues at UPenn addressed these problems by generating mice with an inducible TDP-43 transgene in neurons only. Lacking a nuclear localization signal, the transgenic TDP-43 aggregates in the cytoplasm. After induction of this transgene at five weeks of age, the mice rapidly formed TDP-43 deposits and lost endogenous nuclear TDP-43.
They developed a selective loss of certain motor neurons, and died at 13 to 15 weeks old. That said, when researchers switched off the transgene after six weeks, the mice recovered. The effect was dramatic. Remaining motor neurons re-innervated previously wasting muscle, and the mice lived to a normal age. This work is published (see Nov 2015 conference news; Walker et al., 2015; Spiller et al., 2016).
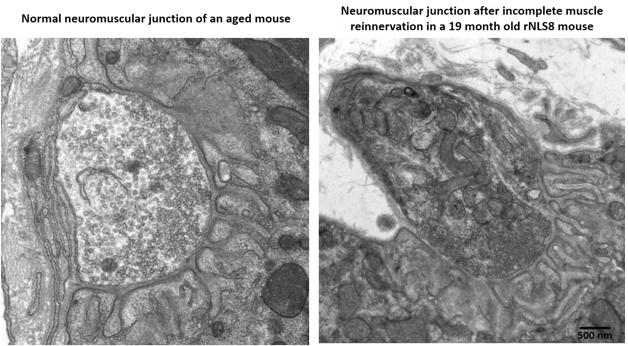
Aged mice recover poorly after a TDP-43 transgene switches off, with partially innervated, disorganized neuromuscular junctions (right) compared to age-matched controls (left). For pictures of recovering young transgenic mice, see upcoming paper. [Courtesy of Krista Spiller and Virginia Lee.]
In Munich, Lee fleshed out this picture with fresh data. She reported that young mice still recovered well even after eight weeks of transgene expression, when they were at death’s door. Her group stressed mice by cycling the transgene on and off every two weeks for six cycles before turning it on permanently. Mice remained healthy during the gene cycling, and followed a normal disease course after the final induction. However, these mice started as adolescents, whereas people do not develop this disease until late in life. To model this, the researchers induced the transgene in middle-aged mice more than a year old. The older mice developed the disease as rapidly as their younger littermates, but they declined faster and died sooner, at four to six weeks after induction. Middle-aged mice tolerated the gene cycling as well as the young ones. But when the TDP-43 transgene was switched off after six weeks, the older mice did not recover as well as young mice did. Surviving motor neurons only partially re-innervated muscle, and their limbs remained weak. Exactly why the older mice recovered poorly is unclear, but Lee noted that their neuromuscular junctions appeared less organized than did those in young, healthy animals after re-innervation (see image above; paper in press).
Researchers were intrigued that TDP-43 had to be permanently active in order to trigger neurodegeneration and death. They said this may help illuminate how the protein damages aging brain. “This model looks promising,” said Manuela Neumann of the German Center for Neurodegenerative Diseases (DZNE). Brad Boeve of the Mayo Clinic in Rochester, Minnesota, called the talk “one of the hottest things” he heard at the conference.
Researchers also want knock-in mouse models of TREM2, the microglial receptor whose variants inflate the risk of FTD, ALS, and AD (see Oct 2012 news; Oct 2013 news; Feb 2014 news). In Munich, Gernot Kleinberger of Ludwig-Maximilians University described the generation of knock-in T66M mice. In people, this mutation lowers levels of the protein; it causes an autosomal recessive early onset dementia when homozygous and predisposes to FTD when heterozygous. Previous studies by senior author Christian Haass at LMU found that this variant becomes stuck inside cells rather than trafficking to the surface membrane (see Apr 2015 conference news).
In keeping with this, Kleinberger reported that the knock-in T66M mice make almost no soluble TREM2, which is produced by cleavage at the cell surface, but do accumulate immature TREM2 inside microglia and bone marrow-derived macrophages. The mice live and make healthy bones, he noted. However, their midbrain develops with a delay and, starting at six months, less blood flow reaches their brains. After one year of age, the mice’s microglia are less activated than those in controls, as seen by PET imaging. In addition, bone marrow-derived macrophages isolated from the knock-in brains poorly phagocytose bacteria and fibrillary Aβ in vitro. Several studies report that TREM2 aids microglial activation and phagocytosis (see Feb 2015 news; Jul 2016 news). Kleinberger is currently collecting behavioral data. Because this model includes many features seen in people with TREM2 mutations, it might be valuable for testing drugs targeting the receptor, he suggested.
Identifying Disease Beginnings
Having better mouse models will enable researchers to investigate how disease begins and progresses. Many scientists now believe that misfolded, pathologic proteins spread along anatomic connections in numerous neurodegenerative diseases, though the concept has also been challenged (see Apr 2015 conference news; Jan 2016 news; Apr 2016 webinar). At ICFTD, speakers added new evidence for this process in FTD, some from animal models and some from postmortem studies.
John Trojanowski described data from his and Lee’s labs at UPenn. Previously, they had reported that injecting either synthetic tau fibrils or pathological tau isolated from AD brain into transgenic tau PS19 mice jump-started tau pathology, with greater verisimilitude to AD-like tangles than seen in aged, uninjected PS19s (see Dec 2014 conference news; Peeraer et al., 2015; Iba et al., 2015). In Munich, Trojanowski reported that injecting tau extracts from AD brain into wild-type mice also gave rise to AD-like tau pathology, although a milder one than in the transgenics. The new tangles were composed of endogenous mouse tau, and spread from the injection site to anatomically connected brain regions. Importantly, injecting AD tau extracts into a tau knockout mouse did not produce pathology. The data confirm that misfolded tau proteins can act as proteopathic seeds and recruit native versions, even at endogenous levels, and they strengthen the idea that tau pathology could propagate in this way through human brain, Trojanowski said. Christian Haass of Ludwig-Maximilians University, Munich, who was not involved in the work, was impressed by this evidence of propagation in a wild-type mouse.

TDP Exodus.
In an FTD patient, TDP-43 (brown) flees the nucleus and gathers in cytoplasmic speckles in VEN, aka spindle cells (long vertical cells). Top and leftmost one show speckles, bottom has normal nuclear TDP. [Courtesy of Bill Seeley.]
But how does disease begin in a person? Bill Seeley of the University of California, San Francisco, described a working model that posits a tug of war between natively folded and misfolded forms of proteins. People have intrinsic regional vulnerabilities to protein misfolding based on their genetics, lifestyle, and other factors. Pathology expresses itself first in the most vulnerable neurons, then leaks out of this epicenter into neighboring cells along network connections, Seeley believes. In behavioral variant FTD, the most vulnerable neurons appear to be von Economo neurons (VENs, aka spindle cells) and fork cells found in the anterior cingulate cortex and frontal insula, Seeley said. He had previously reported that fork cells and VENs selectively die in bvFTD (see Kim et al., 2012).
These cells are large layer 5 projection neurons, similar to the corticospinal neurons that degenerate in ALS, he noted. Regions containing the VENs and fork cells form part of the salience network, which sputters in people with bvFTD. To investigate their vulnerability, Seeley and colleagues analyzed postmortem tissue from 17 people who had had bvFTD and/or ALS, and 10 age-matched controls. They found that more than one-quarter of VENs and fork cells in FTD patients harbored TDP-43 inclusions, while only 8 percent of neighboring layer 5 cells did. Deposits were present in people who had just developed symptoms, and their extent correlated with clinical severity, but were absent in controls. By the time of death, FTD patients had lost up to half of their VENs and fork cells. In surviving cells, dendrites and the cell body had shrunk by a third in cells with TDP-43 inclusions compared to those without. In future work, Seeley wants to find out what renders these neurons especially susceptible to TDP-43.
Other investigators focused on propagation itself. Glenda Halliday of the University of New South Wales, Randwick, Australia, noted that data from human postmortem studies support the concept of protein spread, with distinct patterns in different diseases. Scientists led by Lee and Trojanowski recently proposed four neuropathological stages of TDP-43 pathology in ALS patients, based on the regions affected in people with an increasing pathological load (see Brettschneider et al., 2013). Meanwhile, Halliday and colleagues developed a scheme for differentiating neuropathological bvFTD-TDP from ALS based on deposits in the anterior cingulate cortex versus the hypoglossal nucleus (see Tan et al., 2015). These studies highlight the varied propagation patterns of each disease, Halliday said. In bvFTD, lesions start in frontal regions and move back through the brain, whereas in ALS, inclusions first appear in the motor cortex and then descend into anterior and posterior brain regions. This staging has been replicated by other groups as well (see Fatima et al., 2015; Takeuchi et al., 2016). The data support the idea that misfolded proteins propagate along axons, she noted.
Ultimately, researchers believe that understanding onset and progression will enable them to develop strategies to slow or prevent disease, Seeley said. “I have hope [for treatments] that is born out of the progress made in the last 10 years,” he told the audience.—Madolyn Bowman Rogers
References
News Citations
- Are TDP-43 Mice Living Up to Expectations?
- ALS Model Mice Roar Back When Human Transgene Silenced
- Mutations in TREM2 Cause Frontotemporal Dementia
- Fall Flurry of Letters Kicks Up Dust Around TREM2
- TREM2 Variant Doubles the Risk of ALS
- Microglia—Who Are You Really? New Clues Emerge
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- Protein Propagation Real, but Mechanisms Hazy
- News Brief: More Evidence for Aβ Spread Between People
- New Target Has Legs: Tau PET, Mice, and Antibodies
Webinar Citations
Research Models Citations
Paper Citations
- Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015 Nov;130(5):643-60. Epub 2015 Jul 22 PubMed.
- Spiller KJ, Cheung CJ, Restrepo CR, Kwong LK, Stieber AM, Trojanowski JQ, Lee VM. Selective Motor Neuron Resistance and Recovery in a New Inducible Mouse Model of TDP-43 Proteinopathy. J Neurosci. 2016 Jul 20;36(29):7707-17. PubMed.
- Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E, Van Acker N, Kemp JA, Mercken M, Brunden KR, Trojanowski JQ, Dewachter I, Lee VM, Moechars D. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015 Jan;73:83-95. Epub 2014 Sep 16 PubMed.
- Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC's afferent and efferent connections. Acta Neuropathol. 2015 Sep;130(3):349-62. Epub 2015 Jul 7 PubMed.
- Kim EJ, Sidhu M, Gaus SE, Huang EJ, Hof PR, Miller BL, DeArmond SJ, Seeley WW. Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex. 2012 Feb;22(2):251-9. Epub 2011 Jun 8 PubMed.
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VM, Braak H, Trojanowski JQ. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013 Jul;74(1):20-38. PubMed.
- Tan RH, Kril JJ, Fatima M, McGeachie A, McCann H, Shepherd C, Forrest SL, Affleck A, Kwok JB, Hodges JR, Kiernan MC, Halliday GM. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain. 2015 Oct;138(Pt 10):3110-22. Epub 2015 Jul 31 PubMed.
- Fatima M, Tan R, Halliday GM, Kril JJ. Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol Commun. 2015 Jul 28;3:47. PubMed.
- Takeuchi R, Tada M, Shiga A, Toyoshima Y, Konno T, Sato T, Nozaki H, Kato T, Horie M, Shimizu H, Takebayashi H, Onodera O, Nishizawa M, Kakita A, Takahashi H. Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: Evidence for clinico-pathologic subtypes. Acta Neuropathol Commun. 2016 Jun 23;4(1):61. PubMed.
Further Reading
News
- Indianapolis: Dissecting the Pathways Behind Frontotemporal Dementia
- At 2016 Summit, Field Tackles AD-Related Dementias One By One
- Brain Atrophy Patterns Support Distinct Subtypes of Frontotemporal Dementia
- Paper Alert: Autoimmunity in Another C9ORF72-Deficient Mouse Strain
- New C9ORF72 Mice Develop Symptoms Resembling ALS/FTD
- Unleashed From the Nucleus, TDP-43 Wreaks Havoc in Mitochondria
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.