Mutations
MAPT P301S
Quick Links
Overview
Pathogenicity: Frontotemporal Dementia : Pathogenic
Clinical
Phenotype: Frontotemporal Dementia
Position: (GRCh38/hg38):Chr17:46010388 C>T
Position: (GRCh37/hg19):Chr17:44087754 C>T
dbSNP ID: rs63751438
Coding/Non-Coding: Coding
DNA
Change: Substitution
Expected RNA
Consequence: Substitution
Expected Protein
Consequence: Missense
Codon
Change: CCG to TCG
Reference
Isoform: Tau Isoform Tau-F (441 aa)
Genomic
Region: Exon 10
Research
Models: 7
Findings
This missense mutation is associated with a variable clinical presentation, between, and even within, families.
This mutation was first reported in a Dutch family with early onset frontotemporal dementia with parkinsonism (Sperfeld et al., 1999). The reported pedigree shows eight affected individuals over six generations. Although limited clinical information is available for most family members, the proband and her mother both experienced early changes in mood, behavior, and memory, at age 25 and 30 respectively. Later in the disease course they developed movement disorders (e.g., stereotyped behavior, bradykinesia) and epilepsy. The proband's maternal grandfather was also affected, with symptom onset before age 40. He too had developed seizures, six months prior to his death at age 46. Segregation with disease could not be determined due to lack of DNA from family members other than the proband.
This mutation was also reported in an Italian family with two affected individuals, a father and son dyad (Bugiani et al., 1999). Their clinical features differed and they received different diagnoses: FTD for the father and corticobasal degeneration for the son. Disease onset was early in both, in the third decade of life, and progressed rapidly. Symptoms in the father started with changes in mood, memory, and concentration, followed by rigidity, apathy, visual and auditory, hallucinations and delusions. He died at age 36. Symptoms in the son started with problems moving his hand. He later developed rigidity, dystonia, supranuclear palsies, and myoclonus.
The P301S mutation was also associated with FTD with parkinsonism in an Algerian family of Jewish ethnicity (Lossos et al., 2003). The reported pedigree shows seven affected individuals over three generations. Disease in this family was characterized by early changes in personality and affect with some mild gait deficits. The initial symptoms evolved into progressive cognitive and motor deterioration leading to dementia and severe motor impairment within three to five years. Seizures and myoclonus were not associated with disease in this family. Three affected family members were shown to be mutation carriers, suggesting segregation with disease.
This mutation was also detected in a Japanese man who developed FTD with parkinsonism (Yasuda et a., 2000). His symptoms emerged with a tremor in his hand at age 37. He went on to develop extensive parkinsonism symptoms, including rigidity, postural instability, bradykinesia, and gaze palsy. He developed memory loss and behavioral changes, but was not affected by epilepsy or myoclonus. He died at age 42. Segregation with disease could not be determined due to lack of DNA from family members, but he did have a positive family history of disease. His father developed a gait disturbance at age 36 and died two years later, and his grandfather, who died at age 40, was also symptomatic.
Neuropathology
Neuropathological findings from the Italian mutation carrier with FTD showed frontotemporal atrophy with "knife-edge" atrophy in the fronto-orbital region. Extensive neuronal loss, vacuolation, and gliosis were documented, along with demyelination, spongiosis, and gliosis in the white matter of the prefrontal lobes. Rod-like and semi-circular inclusions were seen in cortical neurons. Pick body-like inclusions were present in neurons of the substantia nigra and thalamus among other brain regions. Extensive hyperphosphorylated tau pathology was observed both neurons and glia (Bugiani et al., 1999).
Biological Effect
Recombinant tau protein with the P301S mutation showed a greatly impaired ability to promote microtubule assembly (Bugiani et al., 1999).
Last Updated: 18 Jul 2024
References
Paper Citations
- Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, Ludolph AC. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999 Nov;46(5):708-15. PubMed.
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999 Jun;58(6):667-77. PubMed.
- Lossos A, Reches A, Gal A, Newman JP, Soffer D, Gomori JM, Boher M, Ekstein D, Biran I, Meiner Z, Abramsky O, Rosenmann H. Frontotemporal dementia and parkinsonism with the P301S tau gene mutation in a Jewish family. J Neurol. 2003 Jun;250(6):733-40. PubMed.
- Yasuda M, Yokoyama K, Nakayasu T, Nishimura Y, Matsui M, Yokoyama T, Miyoshi K, Tanaka C. A Japanese patient with frontotemporal dementia and parkinsonism by a tau P301S mutation. Neurology. 2000 Oct 24;55(8):1224-7. PubMed.
Further Reading
Papers
- Bugiani O. FTDP-17: phenotypical heterogeneity within P301S. Ann Neurol. 2000 Jul;48(1):126. PubMed.
- Morris HR, Khan MN, Janssen JC, Brown JM, Perez-Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN. The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol. 2001 Nov;58(11):1813-6. PubMed.
- Alberts N, Groen K, Klein L, Konieczny MJ, Koopman M. Dorsal root ganglion neurons carrying a P301S Tau mutation: a valid in vitro model for screening drugs against tauopathies?. J Neurosci. 2014 Apr 2;34(14):4757-9. PubMed.
- Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B, Spina S, Baker M, Hutton M, Elder JW, Berger SL, Heflin KA, Hardy J, Momeni P. Characteristics of frontotemporal dementia patients with a Progranulin mutation. Ann Neurol. 2006 Sep;60(3):374-80. PubMed.
Learn More
Protein Diagram
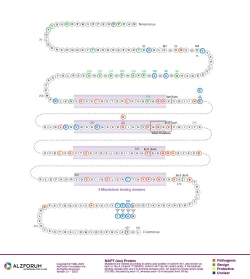
Primary Papers
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999 Jun;58(6):667-77. PubMed.
- Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, Ludolph AC. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999 Nov;46(5):708-15. PubMed.
- Lossos A, Reches A, Gal A, Newman JP, Soffer D, Gomori JM, Boher M, Ekstein D, Biran I, Meiner Z, Abramsky O, Rosenmann H. Frontotemporal dementia and parkinsonism with the P301S tau gene mutation in a Jewish family. J Neurol. 2003 Jun;250(6):733-40. PubMed.
Other mutations at this position
Alzpedia
Disclaimer: Alzforum does not provide medical advice. The Content is for informational, educational, research and reference purposes only and is not intended to substitute for professional medical advice, diagnosis or treatment. Always seek advice from a qualified physician or health care professional about any medical concern, and do not disregard professional medical advice because of anything you may read on Alzforum.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.