Encompassing more than half dozen subtypes, linked to at least four different protein aggregates and at least six distinct genes, frontotemporal dementia would seem like a researcher’s nightmare. And yet, its science is advancing so fast that they have never been more hopeful. Read Tom Fagan’s coverage of the 11th International Congress on FTD, held in Sydney, to catch up on the news.
11th ICFTD Meeting in Sydney Sorts Out Clinical Subtypes
Indigenous Australian people opened the 11th International Conference on Frontotemporal Dementia at the International Convention Center, Sydney, November 11–14. Koomurri performers welcomed more than 600 attendees from 37 countries. Chaired by University of Sydney’s Olivier Piguet on behalf of the local organizing committee, this latest installment of the biannual conference covered the gamut from basic biology to caregiving. A carers' day engaged affected families with personal experiences and updates on the latest research, clinical trials, care planning, and study initiatives. It reinforced, for researchers, the need to better diagnose, track, and treat the subtypes of FTLD. Scientists agreed that key to all three is nailing down what contributes to the cadre of protein pathologies that spread through specific cells and regions of the brain.
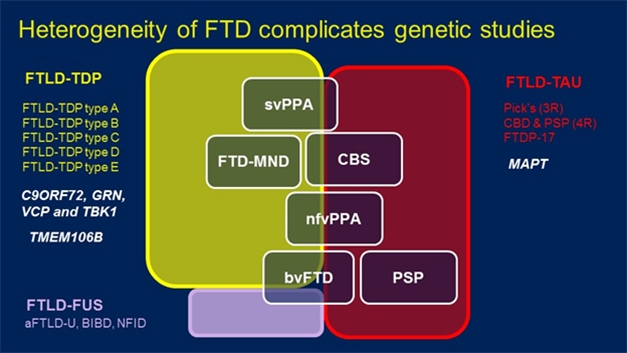
FTD Spectrum. Mutations in a variety of genes (white, left) can lead to pathological aggregates of TDP-43 (yellow), FUS (purple), and in the case of the MAPT gene, tau (red), underlie a handful of clinically distinguishable diseases (white boxes). [Courtesy of Rosa Rademakers.]
It’s complicated, confusing to many, and urgently requires clearer mapping of gene variants to pathologies and, especially, clinical phenotypes. For example, the field currently recognizes five different forms of FTLD based on the morphology of TDP-43 alone. Type A to E FTLD-TDP can manifest as behavioral variant FTD, FTD with motor neuron disease, semantic and non-fluent types of primary progressive aphasia, or corticobasal syndrome. Independently of TDP-43, some of these same clinical diagnoses are made when patients have inclusions of the microtubule-binding protein tau.
Hints at what causes FTD come from genetics. Variants in the tau gene, MAPT, cause the tauopathies, whereas variants in genes encoding four different proteins—C9ORF72, progranulin, valosin-containing protein, and TANK-binding kinase 1—lead to FTD marked by TDP43 inclusions. Could other variants influence what type of TDP43 morphology forms?
Rosa Rademakers at Mayo Clinic, Jacksonville, Florida, has collaborated with dozens of researchers in North America, Europe, and Australia to sequence whole genomes of people with pathologically confirmed FTD-TDP43. From 1,151 samples, the largest collection of FTLD brains to date, Rademakers has thus far sequenced genomes from 517 people with pathologically confirmed TDP43 disease who have no known pathogenic mutations in C9ORF72, PGRN, VCP, or TBK-1.
Comparing these genomes against those from 838 control samples from the Mayo Clinic, the geneticists uncovered two common single-nucleotide polymorphisms on chromosome 7q36 that reached genome-wide significance for association with FTLD. The SNPs lie in intron 1 of the gene for dipeptidyl peptidase like 6. DPP6 was previously linked to FTLD by Rita Cacace in Christine Van Broeckhoven’s group at the University of Antwerp (Sep 2016 conference news). Cacace found an inversion that disrupted the gene. With the new result, Rademakers considers DPP6 a likely candidate gene, though she agreed the data need replication—no small feat in a rare disease.
In the meantime, supporting data come from mice, which exhibit behavioral problems when they lack the gene. How DPP6 functions is unclear, but because it lacks a catalytic serine normally found in serine proteases, this peptidyl peptidase-like protein has no peptidase activity. Instead, DPP6 seems to modulate voltage-gated potassium channels needed for neurotransmission, said Rademakers.
Do any common variants account for specific TDP43 subtypes? Among 162 samples with Type A TDP43, Rademakers’ screen identified rs5848. This SNP lies downstream of the PGRN gene, and Rademakers and others have previously linked it to FTD (Cruts et al., 2006; Baker et al., 2006; Rademakers et al., 2008). The minor T allele seems to reduce progranulin expression. “If you have two copies, you have less progranulin, though not as little as if you have PGRN mutations,” said Rademakers. “This shows that progranulin deficiency may be a cause of FTD not only in people with PGRN mutations,” she said.
Analysis of 184 type B samples turned up a significant hit around UNC13A, another gene previously linked to FTD and ALS (Apr 2018 news; van Es et al., 2009). Scientists believe this protein regulates neurotransmitter release. Rademakers thought it telling that this association emerged from such a small sample set. Alas, in 143 Type C samples, the researchers were unable to find any variants linked with disease.
Next, Rademakers and colleagues looked for rare variants that might explain any form of TDP43 FTD. “We focused on loss-of-function mutations because we had the power to do that,” she said. This identified 61 genes which had at least three mutations among cases versus none in controls. One was the known FTD gene TBK1. ToppGene, an online platform that prioritizes candidates based on functional similarity, ranked three of the other 60 genes—DHX58, TRIM21, and IRF7—the highest. Their proteins perform similar roles to TBK1, being involved in innate immune signaling that leads to production of type I interferons. ToppGene further identified IRF3, IRF8, and NOD2, which are involved in the same pathway. Going back to the rare-variant analysis, Rademakers found one loss-of-function variant in each of these genes among the FTD cases, suggesting they may be candidate FTD genes, as well.
Another rare-variant came from a case-control study by Jennifer Yokoyama at the University of California, San Francisco. Among self-described non-Hispanic white volunteers, she looked for variants enriched in pathology-confirmed FTD cases compared with healthy age-matched controls. She focused on genes with four or more rare SNPs likely to have functional effects. Among 14,459 genes in an initial discovery data set of 62 cases and 2,599 controls, Yokoyama found that, on aggregate, nine variants of the MFSD8 gene occurred in 6.5 percent of FTLD patients but in only 0.4 percent of controls. Deeper analysis of 94 patients and 3,541 controls called the difference significant.
The MFSD8 gene encodes a lysosomal protein. Homozygous mutations at five different amino acids in MFSD8 cause the lipid storage disease late infantile neuronal ceroid lipofuscinosis (Stogmann et al., 2009). In this respect, MFSD8 is uncannily like progranulin, since homo- and heterozygous mutations in PGRN cause childhood lipid storage disorders and FTD, respectively.
Homozygous MFSD8 mutations deplete neurons in layer V of the frontal cortex, a region affected in FTD. In Sydney, Yokoyama reported that in heterozygous carriers with FTD, MFSD8 protein levels were elevated in the frontal gyrus, as were levels of lysosomal markers LAMP-2, CTSD, and LC3-II. This, too, is similar to what is seen in progranulin heterozygotes.
These changes suggest a deficit in lysosomal function, which was apparent in fibroblasts taken from carriers. Further, Yokoyama found that overexpressing one copy of the MFSD8 variant F379S in HEK cells caused small fragments of the protein to accumulate on the cell surface. “This suggests that altered trafficking of MFDS8 to lysosomes could result in reduced protein turnover, consistent with our observations of increased MFSD8 protein levels and reduced lysosomal functioning,” said Yokoyama. Some of this data appeared in the October 31 Acta Neuropathologica (Geier et al., 2018).
Other scientists at the meeting wondered if MFSD8 variants alter levels of progranulin, TDP43, or other proteins implicated in FTD. Yokoyama has not looked at this yet, but said that experiments with fibroblasts from MAPT and GRN mutations carriers hint as much.
Taking a different tack, Edward Lee, University of Pennsylvania, Philadelphia, reported that variants in C9ORF72 might predispose to corticobasal degeneration (CBD). While long hexanucleotide expansions in this gene cause FTD/ALS, Lee wondered if there were pleiotropic effects dependent on expansion length. There is precedent for this: For example, long polyglutamine expansions in ataxin-2 cause spinocerebellar ataxia, while shorter ones increase risk for ALS (Aug 2010 news). Moreover, earlier hints tying C9 expansions larger than 17 repeats to Parkinson’s disease were never proven, said Lee (Nuytemans et al., 2013; Nuytemans et al., 2014). He wondered if a PD mimic, such as CBD, might have been the real McCoy.
To test this idea, Lee analyzed samples from the largest cohort of autopsy-confirmed CBD cases to date, comprising tissue from 355 patients. He genotyped these for C9ORF72 using capillary electrophoresis to quantify expansions sizes, and compared them to expansions among 11,087 controls. Historically, expansion size has been difficult to determine by nucleotide amplification or Southern blot, because the sheer number of hexanucleotide repeats interferes.
Lee reported that repeats smaller than 17 were rarer in CBD patients than controls, but repeats larger than 17 were twice as likely to occur in CBD patients than controls. Why would these relatively small repeats cause CBD while repeats hundreds or even thousands long cause FTD/ALS? Lee believes it’s about gene expression. C9ORF72 has three isoforms, and in the brain samples, mRNA levels for the third isoform rose as expansions grew from 17 to 30 repeats. “This was a surprise,” he said. To corroborate, researchers in his lab used CRISPR to insert intermediate-sized repeats into the C9ORF72 gene in isogenic human neuronal precursor cells. Here, too, these repeats boosted expression of isoform 3.
How would this overexpression cause CBD? Lee does not suspect foci of expanded C9ORF72 mRNA found in FTD/ALS, or poly-dipeptides transcribed from said RNA. Instead, he thinks the intermediate repeats cause tau to aggregate. Using tau biosensor cells pioneered by Marc Diamond, Lee found that seeds from CBD brain tissue precipitated new tau aggregates twice as fast when C9ORF72 was overexpressed.
Others were intrigued. Rademakers has seen expansions affect expression in the same way. She asked how that could relate to CBD, or why it would be specific for CBD. Had Lee looked at expansion sizes in progressive supranuclear palsy? Lee said not yet.
Others wondered about expansions of 30 to100 repeats and about growth of expansions with age, since there is evidence that this happens in somatic cells. Lee said he has no expansions of this size in his sample set, but knows of one person carrying 40 to 50 repeats who was clinically normal but had evidence of C9 RNA foci and dipeptide repeats. At best, Lee thinks such repeat sizes might be a risk factor for CBD. He has found no evidence for expansion growth, but said newer, more sensitive techniques would be worth testing.
Multiple Gene Variants
One of the most puzzling hurdles to understanding FTD is that many of the implicated genes lead to extremely heterogeneous clinical presentations. C9ORF72 expansions are found in patients with FTD, ALS, and even psychosis. What causes such pleiotropy? Mathieu Barbier, Brain and Spine Institute, Paris, wondered if it’s other genetic variants. To get at this, he examined age at onset (AAO) among C9 expansion carriers. While their mean AAO was 60, symptoms can strike as early as 30 and as late as 90.
Barbier looked within families to identify other variants that influence onset. Among 333 affected members of 133 families collected through the the French Reference Centre for Rare and Early Dementias, he found parent-offspring and sibling-sibling AAO correlations that suggested a contribution from a variant on the X chromosome. Taking a different tack, he sorted 75 patients from 34 families into those whose AAO was very similar (within two years) or very different (20-plus years apart). Then, using linkage analysis of 20 concordant and 30 discordant pairs of relatives, he found two suggested links to AAO, one again on chromosome X, and another on chromosome 9. On the latter, the closest gene was PTPRD, which encodes a protein phosphatase found in excitatory presynapses. Lo and behold, the closest gene to the X chromosome locus was SLITRK2, which encodes a postsynaptic protein that turns out to interact with PTPRD in mice.
To further narrow down the loci, Barbier genotyped the concordant and discordant pairs. This turned up 12 SNPs in eight loci, including those near SLITRK2 and three other genes involved in synaptic function—CTNNA2, LRRTM1, and DAAM1. As expected, cross-mapping the linkage and association data revealed one SNP, rs1009776, which lies just upstream of the SLITRK2 gene. This polymorphism associated with age of onset, but only in men. In replicating the analysis in a cohort of 124 unrelated C9ORF72 patients from the International Frontotemporal Dementia Genomics Consortium coordinated by Raffaele Ferrari, University College London, only rs1009776 reached significance and again only in men. Barbier found no association between this SNP and risk for ALS or FTD in people who carry GRN mutations, suggesting the association is specific for C9ORF72 disease. He said work is in progress to understand the functional effect of this variant.
As for GRN mutation carriers, Rademakers is part of a worldwide consortium to study the effect of genetic variants on disease risk and age of onset in this population. To date they have genotyped whole genomes of 382 unrelated symptomatic patients who share 120 different GRN mutations among them. The genotyping revealed no variants that affect AAO, but did turn up variants in two loci that modify risk. These were near the GFRA2 and TMEM106B genes.
Previously, Rademakers and colleagues reported that minor alleles in the TMEM106B gene protect C9ORF72 mutation carriers from disease (van Blitterswijk et al., 2014). In Sydney, she said the gene also protects GRN mutation carriers. “TMEM106B has turned into my favorite gene,” quipped Rademakers, believing it holds the key to a natural cure for GRN mutations. Christian Haass, DZNE, Munich, agreed. “This is super important,” he said. “If we can figure out how this gene protects, then we should be able to treat the disease,” he said.
Based on her data, Rademakers said that a GRN mutation carrier who has one copy of the protective TMEM106B allele is half as likely to get sick while most people with two protective variants never get it. She calculated that 14 percent of the general population is homozygous for the protective TMEM106B variant. “Why do we never see GRN mutation carriers with two copies of this TMEM106B allele in the clinic?” she asked. “Because they rarely get FTD.” She urged the field to debate the merits of genetic testing for TMEM106B in GRN carriers. But in a twist, she also showed how complex the interplay can be between genetic variants. She described one GRN mutation carrier who was homozygous for the protective TMEM106B haplotype; this man did not have FTD but Parkinsonism. It turned out he also carried an R816C variant in the CSF1R gene that has been linked to clinical features of PD. “That is likely what caused the Parkinsonism, not the GRN mutation,” she said.—Tom Fagan
Natural History Studies Provide Foundation for FTD Research
At the 11th International Congress on Frontotemporal Dementia, held November 11–14 in Sydney, no two acronyms were heard more than ARTFL and LEFFTDS. For those readers who have not built them into their FTD vocabulary yet, Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) are closely related, ongoing longitudinal studies of familial and, in the case of ARTFL, also sporadic FTD. Researchers are banking that these two cohorts, and their proposed continuation under the umbrella of a single study called ALLFTD, a.k.a. the ARTFL LEFFTDS Longitudinal FTD study, will chart onset and progression of the main types of FTD, and in the process build well-characterized, trial-ready cohorts for future therapeutic studies. ARTFL and LEFFTDS began in 2014, garnering the biggest grant the NIH ever gave to FTD research (Nov 2014 conference news). Where do they stand now?
Leah Forsberg, Mayo Clinic, Rochester, Minnesota, reviewed LEFFTDS, while Adam Boxer of the University of California, San Francisco, laid out ARTFL progress. Bradley Boeve, also at the Mayo in Rochester, and Howard Rosen of UCSF co-lead the NIH-funded LEFFTDS, while, as part of the larger Rare Diseases Network, the ARTFL consortium is coordinated by Rosen, Boxer, Hilary Heuer, and others at UCSF. Both studies share a similar design and even subjects, with all but four LEFFTDS participant also enrolled in the ARTFL familial FTD cohort, Heuer told Alzforum. The two studies collect the same types of biological data and store it in a common database. They both aim to track rates of decline and look for genetic variants and biomarkers that modulate disease onset and progression.
Forsberg reported that seven centers in the U.S. and one in Canada had recruited 367 volunteers from 65 families into LEFFTDs as of late October. Almost all are Caucasian, seven are Asian, and 10 represent all other non-Caucasians. Fifty-five percent are women. Volunteers range from 35 to 63 years old and have about 15 years of education on average. About 129 healthy participants carry none of the known FTD mutations and show no signs of FTD. About the same number carry a mutation and are asymptomatic, while 110 have a mutation and are asymptomatic.
Clinical Diagnoses. The range of diagnoses among ARTFL volunteers with sporadic FTD. [Courtesy of Adam Boxer.]
LEFFTDS had hoped to recruit equal numbers of people from families carrying mutations in the tau gene MAPT, the progranulin gene GRN, or expansions in the C9ORF72 gene. They pretty much succeeded, recruiting 26, 28, and 33 percent of the cohort from such families, respectively. Ten distinct MAPT and 18 distinct GRN mutations are represented in the cohort. Of 145 people from families carrying C9ORF72 hexanucleotide expansions, two people had both an expansion and a progranulin mutation. Forsberg said that because the genotype data is de-identified, LEFFTDS researchers do not know an individual’s mutation status unless that person reveals it.
To date, all participants have had one clinical visit. Seventy percent have already had a second, 52 percent have had a third, and 16 percent a fourth. About 90 percent of participants have volunteered DNA, RNA, and plasma/serum; have undergone MRI imaging; and have donated peripheral blood mononuclear cells, which can be used to make induced pluripotent stem cells to study the effects of the various mutations on cell biology. Only about 45 percent have agreed to donate cerebrospinal fluid; this is not unusual among North Americans who seem more anxious about lumbar punctures than Europeans. All were evaluated clinically and took a battery of neuropsychological tests.
For its part, ARTFL has recruited 608 people who have sporadic FTD. Among them, 184 had behavioral variant FTD, 128 with progressive supranuclear palsy, 76 with corticobasal syndrome, and 77 and 62 with semantic and non-fluent variants of primary progressive aphasia (see image above). Eighteen had FTD-ALS. As in LEFFTDS, participants were predominantly Caucasian, with a similar number of men and women except for the bvFTD and FTD-ALS patients, of whom 72 and 61 percent, respectively, were men. Of the 608, researchers concluded that 62 did not have FTD but another disease, such as Alzheimer’s, Parkinson’s, or dementia with Lewy bodies.
ARTFL Diagnoses. The breakdown of clinical diagnoses among ARTFL FTD mutations carriers. [Courtesy of Adam Boxer.]
In addition to the 608 people with sporadic FTD, 531 people from familial FTD kindreds have also enrolled in ARTFL. These are families who carry a known FTD mutation, including 363 of the people enrolled in LEFFTDS, or who report a family history that indicates an autosomal-dominant cause. Ninety-two people fell into the latter category, while 184, 104, and 132, came from C9ORF72, GRN, and MAPT families, respectively. Only 19 people had other variants, including those in the VCP and TARDBP loci. Among the 531 recruits, 333 show no symptoms, 84 have mild symptoms, and 114 have full-blown disease, meaning a CDR of 1.0 or greater. Like LEFFTDS, they are predominantly Caucasian and 56 percent are women. Ages ranges from 31 to 70.
ARTFL collects the same biological samples as does LEFFTDS, except no CSF. Among ARTFL volunteers with fFTD, 317 have completed a one-year follow-up visit. Most showed no change in clinical status; 16 went from asymptomatic to mildly impaired. Of those who had symptoms at baseline, seven seemed to improve, meaning they went from CDR 0.5 to CDR 0, while 23 worsened.
Different Cause—Same Pattern. Compared with 30 healthy controls (not shown), people with familial and sporadic bvFTD show similar brain atrophy patterns. [Courtesy of Adam Boxer.]
Many of the participants in ARTFL/LEFFTDS have not been seen often enough for researchers to reach consensus about deterioration among the various subtypes of FTD; still, some trends are beginning to emerge. On a poster, Boxer directly compared sporadic and familial behavioral variant FTD in the ARTFL cohort. He reported that people with MAPT mutations were younger at diagnosis than those with any other mutation or with sporadic bvFTD. This might imply MAPT mutations cause a more aggressive disease, yet Boxer saw little difference in prevalence of neuropsychiatric symptoms among the volunteers. If anything, those with sporadic bvFTD were more likely to be reported by family members or carers as being depressed and irritable. Clinicians also reported more irritability among sporadic bvFTD patients as disease progressed, but found no neuropsychiatric differences between familial and sporadic bvFTD on the first visit to the clinic. MRI data supports the idea that sporadic and familial bvFTD are similar. Atrophy patterns among 23 sporadic and 31 familial patients were all but indistinguishable (see image above), at least for this behavioral variant form. This might be good news for clinical trials, because it suggests that the patients could be combined, making it slightly easier for clinicians to recruit people with this rare disease. For more on outcome measures that might be used in such trials, and on heterogeneity of atrophy patterns among FTD patients in general, see Part 3 of this series.
Going forward, ARTFL/LEFFTDS researchers hope to follow all these patients for at least another five years under the auspices of the ALLFTD study. Rosen told Alzforum that ALLFTD will recruit new participants also. ALLFTD plans to test the predictive value of polygenic risk scores in addition to biomarkers. “We are taking advantage of new research developments and hope to incorporate other data, such as whole genome sequencing, through collaboration with other efforts,” Rosen said.—Tom Fagan
Tracking Onset and Progression of Frontotemporal Dementia
Now that initiatives such as ARTFL and LEFFTDS have gathered sufficient numbers of participants for scientists to tackle prevention and treatment trials (see Part 2 of this conference series), the need for better tools to track onset and progression of disease has become pressing. At the 11th International Congress on Frontotemporal Dementia, held November 11–14 in Sydney, scientists proposed clinical and imaging tests that might be used to monitor changes early in disease.
Bradley Boeve, Mayo Clinic, Rochester, Minnesota, debuted a Multidomain Impairment Rating score designed to broaden the current standard, the FTLD Clinical Dementia Rating scale, so that it accounted for the phenotypic heterogeneity of FTLD. Adam Staffaroni from University of California, San Francisco, outlined NIH EXAMINER, a composite of executive function tests that seems to detect deficits earlier in disease and even in asymptomatic individuals. Along the same vein, Howard Rosen, also from UCSF, reported how an atrophy-based dementia risk score can predict disease onset in FTD mutation carriers.
Tracking Progression.
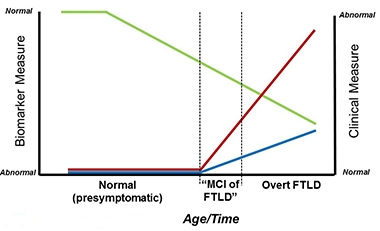
Researchers hope the MIR (blue) and MIR-SS (red) will complement biomarker measures (green) to detect the MCI phase of FTLD. [Courtesy of Bradley Boeve.]
The Multidomain Impairment Rating (MIR) scale builds both on the Clinical Dementia Rating (CDR) scale, which was devised to track clinical and functional decline in dementia patients across etiologies, and on the FTLD-CDR, which expanded the CDR to an eight-item score by adding language and behavior domains not typically affected in Alzheimer’s disease (Knopman et al., 2008). As Boeve said in his talk in Sydney, members of the ARTFL/LEFFTDS Consortium wanted to build a scale for the early phase of FTLD that is akin to the mild cognitive impairment phase of AD. This is when symptoms are so subtle that clinicians find it difficult to diagnose FTLD based on clinical tests alone. The trouble is that because the FTLD-CDR lacks visuospatial, motor, or neuropsychological components, it misses FTLD subtypes such as corticobasal syndrome, Richardson syndrome (a common form of progressive supranuclear palsy), and FTD with parkinsonism or motor neuron disease (MND).
Boeve and colleagues adapted the CDR/FTLD-CDR to encompass known functional and clinical features of FTLD. It integrates data from three sources: the patient; an informant, who is usually a spouse or immediate caregiver; and from neuropsychological tests. Like the CDR, the MIR scale ranges from zero to 3, with 3 being most impaired. The global score works like the CDR, while a summary score, the MIR-SS, works like the CDR sum of boxes to provide a more granular assessment of all sub-domains combined.
Boeve and colleagues have been testing MIR for reliability for more than a year. Researchers at Mayo Clinic and at UCSF used the scale to rate 20 ARTFL/LEFFTDS participants at each site. They comprised normal controls and those with mild cognitive and/or behavioral changes typical of the various forms of FTD, including behavioral variant, primary progressive aphasia, and FTD with parkinsonism or MND. Clinicians from both sites assessed all the volunteers and were blinded to results from the other institution. How did the results compare? Though the majority of participants were only mildly impaired, Boeve reported excellent inter-rater agreement between the two sites. Each gave 19 volunteers an MIR score of zero, 10 a score of 0.5, and six a score of 1.0 or 2.0. Overall, 35 volunteers received exactly the same score at each site. The pattern was similar for the MIR-SS. Both ratings scored a Cohen’s kappa coefficient of 0.83 for inter-rater reliability, which most statisticians consider excellent, said Boeve. Additional analyses are planned to assess the MIR.
Boeve said researchers hope to use the MIR in ALLFTD (ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration).
As for longitudinal analysis, Boeve showed how the global and MIR-SS tracked with progression in two people who carried mutations in the tau gene. In both the volume of the frontotemporal lobe, as judged by MRI, had declined prior to onset of symptoms and both scales ticked up from zero at year of onset and as atrophy worsened.
On atrophy and disease progression, Rosen emphasized the need to identify those familial FTD mutation carriers who are within two to three years of becoming symptomatic, so they can be invited into trials. “These are the people we need to study in order to demonstrate that a treatment delays onset of symptoms,” he said. He noted two major obstacles. First, age of onset varies dramatically even among people with the identical mutation. This is true for the tau gene MAPT, the progranulin gene PGRN, or in C9ORF72, which together hold the bulk of familial FTD mutations. Second, since FTD spans heterogeneous pathologies, patterns of atrophy can vary widely. “We had to recognize that different people [with the same mutation] will have symptoms because of different patterns of brain atrophy,” Rosen told Alzforum. “So we designed an analysis to be sensitive to any pattern than might lead to symptoms.” The answer, said Rosen, was to use individualized atrophy maps.
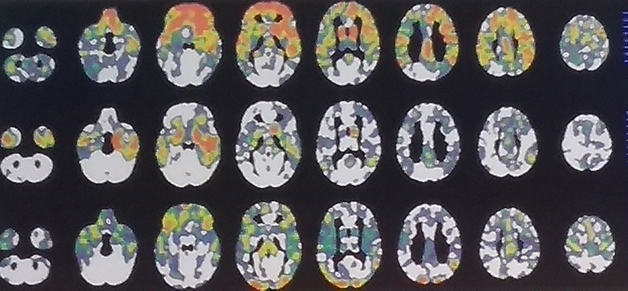
Same Cause—Different Pattern. These three C9ORF72 carriers each are at FTLD-CDR 1, yet their regional patterns of gray-matter shrinkage differ greatly. [Courtesy of Howard Rosen, UCSF.]
To generate these, Rosen quantified each volunteer’s brain volume voxel by voxel using w scores. A w score is like a composite z score but accounts for a host of other co-variables such as age, sex, total intracranial volume, and type of scanner used. The researchers counted voxels that had shrunk by more than two standard deviations compared with normal. Then they tallied those scores across brain regions for people who did, and did not, have dementia to determine how much burden, in which areas, is predictive, and used that in an overall score of total atrophy burden. This burden, regardless of brain region, might be most informative, Rosen argued.
How would this method pan out? Rosen tested it by comparing 311 reference controls with 46 people carrying familial FTD mutations in MAPT, GRN, and C9ORF72. Twenty had an FTLD-CDR score of 1 or higher, while 26 had a zero. Rosen also tested 22 FTLD-CDR 0.5 mutation carriers and 115 non-carrier relatives.
Cross-sectionally, carriers with CDR greater than 1 had a higher atrophy risk score than noncarriers or carriers with CDR zero. The atrophy burden in CDR 0.5 carriers was more variable, and no clear difference from noncarriers emerged. Longitudinally, however, Rosen found that a person’s score did predict dementia. Among 14 FTD mutation carriers who progressed from CDR zero or 0.5 over an average of 1.5 years, a participant’s odds of getting dementia increased by 1.51 for every 0.1 unit increase in atrophy risk score. Rosen calculated that an atrophy risk score of 0.5 (1.0 being the worst) was a cutoff. More than 50 percent of people with scores of 0.5 or higher had developed dementia within two years. “The nice thing about the risk score is that we developed the model based on patterns of degeneration that suggest dementia is oncoming, and then tested it in people who were observed to convert to dementia, and it did improve the ability to predict it,” Rosen said.
Rosen acknowledges that more refinement is needed using larger, independent cohorts, but believes the method could help stratify patients for recruitment into trials and for follow-up analysis. Others wondered if including white-matter changes could improve accuracy. “We certainly have room for improvement,” he agreed, “here we just tried to prove the principle.” Others noticed that mutation carriers who scored above the atrophy cutoff of 0.5 deteriorated quickly, with some being diagnosed with dementia within a year. “Are they the people we want to target in a trial, or is it too late for them to have an intervention?” one audience member asked. Rosen acknowledged that there is not enough information to answer this question. “The score is a probability estimate, so you would have to power your study based on what percentage of patients you expect to progress,” he said. “My hope is that people at that stage, with few symptoms and no problem with daily function, will benefit from drugs.”
Staffaroni and colleagues also used a composite score to estimate progression, but based it on executive function. They, too, hope the composite will circumvent the problem of heterogeneity. They used the NIH-EXAMINER, which was developed at USCF for tracking executive function in clinical trials. It includes measures of working memory, word fluency, and cognitive control, i.e., set shifting, or the ability to multitask, and has detected early cognitive changes in other neurodegenerative diseases, including Huntington’s (Possin et al., 2013; You et al., 2013). “We are looking to measure change in presymptomatic stages, something that might be used in early clinical trials. The CDR and CDR-sb are insensitive to some of the earliest changes” said Saffaroni. The scientists tested the NIH-EXAMINER in parallel with Trails B, a commonly used test of executive function.
Staffaroni recruited subjects from 18 ARTFL/LEFFTDS sites, including 93 FTLD mutation carriers evenly split among MAPT, GRN, and C9ORF72 variants, and 78 noncarriers. From each, 64 had an FTLD-CDR score of zero (69 and 83 percent, respectively), while the remainder were at 0.5. At baseline, carriers scored worse than noncarriers on EXAMINER but no different on Trails B. After one or two follow-up visits, carriers deteriorated more on the EXAMINER than did noncarriers, whereas Trails B again picked up no difference between the two.
Would the differences hold in those who were, at least as measurable currently, asymptomatic? The data suggested yes. When Staffaroni limited analysis to people who were FTLD CDR zero when they entered the study and were tested twice within 1.5 years, those with higher CDR-sb performed worse on the EXAMINER. Furthermore, decline in EXAMINER scores tracked with shrinkage of the frontal, temporal, and parietal lobes.
Staffaroni thinks the EXAMINER might be more sensitive than the CDR-sb, and better suited as a clinical measure for early stage FTD, though he agreed this needs more testing. He also plans to compare if its composite or sub-domain scores work better, and study both in people with specific mutations. “Our sample set included people with mutations in C9ORF72, MAPT, and progranulin, but trials will likely target carriers of a single gene,” he said.—Tom Fagan
You SC, Geschwind MD, Sha SJ, Apple A, Satris G, Wood KA, Johnson ET, Gooblar J, Feuerstein JS, Finkbeiner S, Kang GA, Miller BL, Hess CP, Kramer JH, Possin KL.
Executive functions in premanifest Huntington's disease.
Mov Disord. 2013 Dec 27;
PubMed.
A Proteomics Dive into Cause of Frontotemporal Dementia
If DNA makes RNA makes protein, then genomic variants make RNA make protein … make disease. While scientists have found polymorphisms that associate with various forms of frontotemporal dementia, deciphering how they perturb proteins is proving more complicated. “It’s time we start thinking how to get beyond genetics to a functional understanding of FTD,” said Raffaele Ferrari, University College London, at the 11th International Congress on Frontotemporal Dementia, held November 11–14 in Sydney. Ferrari and others championed the idea that studying co-expression patterns and protein-protein interactions will not only lay bare the underlying biology of FTD, but also identify disease markers and therapeutic targets.
At the conference, some researchers described functional protein modules related to progranulin insufficiency that suggest early inflammatory and lysosomal changes in FTD. Mutations in the progranulin gene are a major cause of familial FTD, while levels of the protein drop in people with sporadic forms as well. Others showed that progranulin-related changes can be cell-specific, tracking one way in microglia and the opposite way in whole brain. They also made a much-sought connection between two key players in the pathology of FTD and amyotrophic lateral sclerosis (ALS)—the lysosome and RNA granules. Last but not least, scientists from Alector, a small startup in South San Francisco, reported that their therapeutic antibody targeting sortilin, a receptor that promotes lysosomal degradation of progranulin, boosts levels of the protein in mice and monkeys and rescues behavioral deficits in the former. AL001 has entered clinical trials.
FTD Modules. A representative model from co-expression analysis places genes for tau (MAPT) and progranulin (GRN), in a network that regulates DNA transcription. MAPT and GRN are colored pink, while genes directly connected to them are blue. In this module, MAPT was a hub. [Courtesy of Ferrari et al., Molecular Neurodegeneration.]
Modules and Cells
For his part, Ferrari capitalized on data from the International FTD Genomics Consortium in his search of gene network changes that point to molecular pathways gone awry in FTD. Using transcription microarray analysis of frontal and temporal cortices from 101 samples from the U.K. Human Brain Expression Consortium, Ferrari uncovered networks of genes that co-express with known FTD genes, including MAPT, GRN, C9ORF72, VCP, OPTN, and TMEM106B. These co-expression analyses turned up modules of genes involved in DNA protection and transcriptional regulation, immune function, lysosomal activity, proteostasis, and the ubiquitin proteasome pathway (Ferrari et al., 2016).
How do these modules function? For that, Ferrari turned to proteomics. He devised a layered approach to build protein interactomes. First, he used products of FTD genes, such as tau and progranulin, as seeds in a weighted protein-protein interaction network analysis. He then used nodes from those protein networks as seeds in subsequent analysis (Ferrari et al., 2017). Putting it all together, he ultimately identified blocks of proteins linked to FTD genes that are involved in DNA repair, cytokine-mediated immune responses, the ubiquitin proteasome system, and the unfolded protein response. “We thought it quite supportive, from disease-mechanism and functional perspectives, that the protein-protein interaction and expression networks, which are both based on Mendelian and GWAS hits, substantially overlaid each other,” he said. The overlap was highest for DNA repair and cellular waste disposal. Among these were four interactome hubs, which were defined as proteins common to at least eight interactomes. One was EP300, a histone acetyltransferase that Ferrari considers a potential biomarker or therapeutic target.
At the conference, others asked if this analysis can pick out what wayward pathway or molecule kick-starts the disease process. Ferrari thinks not. “The more pressing point is that we have done this work in silico, so we need to validate experimentally,” Ferrari said. “We have some of that data because the protein-protein interactions have been determined biochemically, but still need further and more tailored work in FTD models.”
Scientists are using animal models to identify the protein networks and interactomes germane to FTD. The labs of Thomas Kukar,Nicholas Seyfried, and Malu Tansey at Emory University, Atlanta, have compared the brain proteomes of progranulin knockout mice and people with progranulin mutations to find common pathways that might be pathogenic. In the mice, a deep proteome analysis quantified 8,000 different proteins and found differences between Grn KOs and wild-type controls, said Kukar. In three-month-old knockouts, 29 proteins were upregulated over age-matched wild-type, and 26 were downregulated. The proteins included cathepsins B, D, and Z, TPP1, HexA and B, and glial proteins such as GFAP and complement C1q, pointing to lysosomal dysfunction and glial activation. “Lysosome dysregulation and inflammation have been linked to FTD before, but these mice are only three months old, so we think such changes occur earlier than expected and may serve as novel biomarkers of disease progression,” said Kukar. To his mind, this also suggests a subtle disconnect between proteome and transcriptome changes, since the latter are not seen in such young animals. This hints that disease might start with protein-level changes that then cause transcriptional dysregulation.
In 19-month-old mice, the number of upregulated proteins had grown to 119, while the number of downregulated proteins was about the same as in the younger mice. The same lysosomal and microglial proteins were still differentially expressed at that age, but so were new glial markers—complement C4 and S100a4.
Since FTD GRN mutations are heterozygous, how would these changes lead to human disease? By immunohistochemistry, Kukar showed that Grn-heterozygous mice had upregulated the lysosomal activation CD68 by 12 months, and even more by 24 months of age. “We think lysosomes are dysfunctional in Grn heterozygotes, they just need to be older than full knockouts for changes to manifest,” said Kukar.
Like Ferrari, Kukar analyzes correlation networks to probe functional consequences of proteome changes. He identified 29 protein modules in mice. Four most strongly correlated with knockout status and age. To relate this to FTD, he compared the mouse modules with human ones identified in frontal cortex samples from 11 age-matched controls, seven GRN-FTLD cases, and 10 non-GRN cases (Chen-Plotkin et al., 2008). He found 10 modules that overlapped between the mouse Grn knockouts and the familial and sporadic FTD cases.
In Sydney, Kukar focused on a common human/mouse module that was enriched in microglial proteins, complement factors, and proteins found in peripheral immune cells. “This might be evidence that peripheral cells have entered the brain in FTD,” he said, adding that the Tansey lab is now using flow cytometry to measure how many microglia and peripheral immune cells there are in the brain samples. In mice, flow cytometry indicates older Grn knockouts have fewer microglia in the brain than do age-matched wild-type animals. “This was surprising, because the literature would suggest they should have more,” Kukar said. It turns out that peripheral monocytes might make up for the shortfall. Based on cell-surface expression of CD45 and CD11b, the scientists found a strong infiltration of monocytes into Grn knockout brain. Whether the same happens in FTD remains to be seen. As a start, Tansey and colleagues are measuring changes in peripheral blood monocytes from volunteers in the ARTFL/LEFFTDS natural history studies. The data could have important implications for therapeutic strategies in FTD caused by GRN haploinsufficiency, Kukar said.
Looking to Lysosomes
This idea of changes to immune cells in FTD drew further support from Anja Capell, Ludwig Maximilians-University, Munich. Capell also uses mice, but studies directly how deleting GRN affects lysosomal function. Previously, she reported that in 22-month-old mice and mouse embryonic fibroblasts (MEFs), progranulin deficiency results in higher expression of the lysosomal proteins cathepsin B and D, LAMP1, and saposin D. This led to more maturation and activation of cathepsins, which are initially made in an inactive prepro form (Götzl et al., 2014).
Cell-Specific Responses.
Without progranulin, lysosome activity (yellow-red color scale) increases with age in neurons (green), perhaps to compensate for faltering lysosomes in microglia (blue). [Courtesy of Anja Capell.]
Does this cathepsin boost square with lysosome dysfunction in FTD? After all, the lysosome is there to degrade proteins. To investigate, Capell and colleagues used S35 pulse labelling to check protein degradation in MEFs. Indeed, MEFs from GRN knockouts degraded lysosomal substrates, in this case the amyloid precursor protein and its C-terminal fragments, faster than did wild-type MEFs. Capell also saw an age-related uptick in expression of lysosomal membrane proteins such as Cd68 and Ctsd in GRN knockout brain, again pointing to up-, not down-, regulation of lysosomes.
Things started to make sense when the researchers specifically examined microglia they captured from brain extracts using microbeads coated with antibodies to the microglial cell surface marker CD11b. In contrast to whole-brain extracts from GRN knockouts, the microglia did downregulate expression of cathepsin D, and suppressed maturation of cathepsin B. In short, Capell believes that without progranulin, lysosomes become dysfunctional, particularly in microglia, and that this leads to enhanced lysosomal activity in other brain cells.
Others wondered why microglia seem to be persistently activated in older GRN knockouts. Capell suggested this might be the brain’s attempt to compensate for reduced lysosomal activity.
Taking yet another proteomic tack, Michael Ward from the National Institutes of Neurological Disorders and Stroke, Bethesda, Maryland, forged a long-anticipated connection between lysosomes and RNA granules. The latter are hotbeds for accumulation and, potentially, aggregation of RNA-binding proteins that build up in neurons in FTD and ALS, such as TDP-43, FUS, and hnRNPA1. As detailed in a poster, Ward and colleagues, including co-first authors Michael Fernandopulle in his lab, and Ya-Cheng Liao in Jennifer Lippincott-Schwartz’s group at the Howard Hughes Medical Institute, Ashburn, Virginia, used proximity-labelling proteomics to identify proteins that bind to lysosomes. The scientists tied those organelles to axonal transport of RNA granules, which may go awry in ALD/FTD. Meeting organizers awarded the poster first prize in the category for investigators who are early in their career.
In proximity labelling, researchers engineer a protein, lysosomal LAMP1 in this case, to incorporate an ascorbate peroxidase (APEX) unit (Rhee et al., 2013). Add a little hydrogen peroxide and biotin to cells expressing LAMP1-APEX, and presto, it biotinylates nearby proteins, which can then be tracked or isolated using streptavidin. In this manner, Fernandopulle identified 99 different proteins that interact with lysosomes in neurons derived from patient-induced pluripotent stem cells. Comparing those with hits from proximity-labelling of RNA stress granules reported earlier this year by researchers in Gene Yeo’s lab at University of California, San Diego (Markmiller et al., 2018), Fernandopulle found six proteins in common. One was none other than annexin A11 (ANXA11), a vesicular-trafficking protein that is mutated in some ALS patients (May 2017 news; Liu et al., 2018; Tsai et al., 2018).
Ward explained that RNA stress granules use ANXA11 to hitch a ride on endolysosomes as they move through axons. ANXA11 contains a low-complexity N-terminal domain predicted to undergo liquid-liquid phase separation, suggesting the protein blends into the liquid droplets that constitute RNA granules. Its C-terminal contains repeats that interact with membrane lipids and calcium. In collaboration with Peter St George Hyslop’s group in Cambridge, England, Ward showed that ANXA11 undergoes liquid-liquid phase separation and that it interacts with both RNP stress granules and lysosomes in living cells. The ALS-linked ANXA11 mutations D40G, R235Q, and R346C all disrupted phase separation of stress granules and reduced lysosomal binding.
Using a light-activated form of ANXA11 and time-lapse photography, Liao showed that annexin A11 brings lysosomes and RNP granules together and that they travel as one in axons of rat neurons. Again, ALS mutations scupper this interaction and reduce co-trafficking of RNA granules and lysosomes. All told, the work ties RNA granules to lysosomes via ANXA11, and suggests that damage to this interaction contributes to the pathophysiology of both ALS and FTD, said Ward.
A Lysosome-Based Immunotherapy
Showing how the study of protein-protein interactions can pay dividends, Michael Kurnellas updated the audience on Alector’s anti-sortilin program. The company targeted sortilin-1 because mice lacking this sorting receptor produce more progranulin and have no obvious behavioral or lysosomal phenotype (Nov 2010 news on Hu et al., 2010). The researchers developed aSORT1, a.k.a AL101, a monoclonal antibody that binds sortilin-1 with high affinity and prevents progranulin from docking with the receptor. In both wild-type and GRN+/- heterozygous mice, the antibody all but abolished sortilin-1 in white blood cells, while boosting plasma and CSF levels of progranulin about six- and twofold, respectively. Kurnellas did not disclose how much antibody was administered, or for how long. He did report that the same antibody slashes levels of sortilin in cells from cynomolgus monkeys, while boosting serum and CSF levels three- and twofold, respectively.
Does this help? To find out, Kurnellas and colleagues collaborated with Erik Roberson at the University of Alabama, Birmingham. Andrew Arrant in the lab had reported that progranulin-heterozygous mice are a bit of a pushover when they meet another mouse face-to-face in a small tube. They are much more likely to retreat. However, a weekly regimen of aSORT1 made 21-month-old heterozygotes more likely to hold their ground than those treated with a control IgG antibody.
This past August, the U.S. Food a,nd Drug Administration granted orphan drug status to AL001, a humanized monoclonal version of aSORT1. INFRONT, a single-ascending-dose Phase 1 trial began in September. It is slated to recruit 60 healthy volunteers at seven sites in North America and Europe. Kurnellas said a multidose trial in FTD patients carrying progranulin mutations will follow once Phase 1 has completed. INFRONT is expected to finish by March 2020.—Tom Fagan






Comments
No Available Comments
Make a Comment
To make a comment you must login or register.