Paper Alert: Trafficking Variants Cause BACE Buildup in Axons
Quick Links
Does Aβ cause the axonal dystrophies found in the Alzheimer’s disease brain, or do dystrophies cause the Aβ? In the November 18 Science Translational Medicine, researchers led by Giuseppina Tesco at Tufts University, Boston, claim it is the latter. They report that knocking out the transport protein GGA3 in mice disrupts BACE1 movement in neurons, causing axons to swell. When the researchers suppressed GGA3 in a model of amyloidosis, axon dystrophies bloomed before Aβ plaques could be detected, suggesting the atrophies come first. Inhibiting BACE soothed the dystrophies, and curiously, restored BACE trafficking. To cap it all off, the researchers uncovered a loss-of-function variant in GGA3 that associated with late-onset AD, suggesting this whole scenario might accelerate disease. Alzforum reported on some of this data previously (Oct 2016 conference news).
- GGA3 variants stall BACE and cause axon swelling.
- These dystrophies start to produce Aβ.
- GGA3 loss-of-function mutation linked to familial AD.
GGA3, short for Golgi-localized γ-ear-containing ARF binding protein 3, recycles unwanted proteins, including BACE, by shuttling them to lysosomes (June 2007 news). Previously, Tesco found that knocking out GGA3 in neurons halted BACE transport, allowing the protease to accumulate in swollen axons that looked like dystrophic neurites, a hallmark of early stage AD (Tesco et al., 2007). GGA3 knockout also worsened plaque pathology (Oct 2016 conference news). Tesco and colleagues wondered if Aβ caused dystrophic neurites in AD by blocking GGA3 trafficking, or if trafficking problems led to the axonal dystrophies and then to ramped up cleavage of APP and Aβ production.
To find out, first author Selene Lomoio and colleagues turned to confocal microscopy to track the movement of BACE1 and GGA3. In cultured GGA3 knockout neurons that overexpressed BACE1, the protease accumulated in axons, never making it to lysosomes. These knockout neurons had fewer BACE1 vesicles moving either up or down axons, and the vesicles were sluggish. Stationary BACE1 vesicles also accumulated in swollen axons, resembling axonal dystrophies.
In vivo, immunofluorescence and immunohistochemistry revealed more BACE1 in the hippocampi of 4-month-old GGA3 knockout mice than in age-matched wild-type mice (see image below), particularly in the mossy fibers of the dentate gyrus.

BACE Buildup. BACE1 (green) accumulates in GGA3 knockout mouse hippocampus (bottom). [Courtesy of Lomoio et al., Science Translational Medicine, 2020.]
“This exciting study provides compelling evidence that GGA3 deficiency causes BACE1-enriched axonal swelling,” Riqiang Yan at the University of Connecticut Health, Farmington, told Alzforum. He wondered if other AD-related molecules, such as APP, gather at these swellings.
What if GGA3 was reduced in a mouse model of amyloidosis? Lomoio crossed 5xFAD mice with GGA3 knockouts, then examined 2-month-old offspring. At this age, 5xFAD mice have not yet developed cortical Aβ plaques (Oakley et al., 2006). Still, the animals had accumulated BACE1 in axons, which swelled. This suggests that BACE transport faltered and axonal dystrophies grew independently of plaque formation.
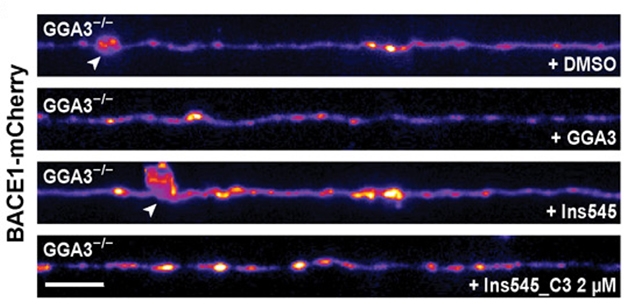
What happens when BACE is blocked? The researchers treated BACE1-overexpressing GGA3 knockout neurons with the β-secretase inhibitor C3. This restored BACE1 trafficking, preventing its accumulation in dystrophies (see image below). To test this in vivo, Lomoio treated 2-month-old GGA3 knockout mice with the brain penetrant BACE MBi-3. The highest dose of 30 mg/kg per day preserved hippocampal axon morphology and reduced Aβ40 levels by more than 75 percent. Taken together, the findings suggest that inhibiting BACE restored its trafficking and prevented axon swelling.

BACE Inhibitors to the Rescue. BACE transport along axons (top panel) falters in GGA3 knockout neurons, leading to axonal swellings (arrow, second panel). Adding GGA3 (third panel) or BACE inhibitors (fourth panel) rescued transport and abolished axon swelling. [Courtesy of Lomoio et al., Science Translational Medicine, 2020.]
Stefan Lichtenthaler at the German Center for Neurodegenerative Diseases (DZNE), Munich, was intrigued by these findings. “This suggests the local production of Aβ contributes to the axonal damage,” he said (see full comment below). Cláudia Almeida at University of Lisbon, Portugal, was surprised the authors did not discuss the role of intracellular Aβ in more depth. “If BACE1 is stalled and accumulates in the axonal swellings, I predict that there will be more Aβ in these swellings even if in vivo extracellular Aβ is not changing,” she wrote (see full comment below).
Since people carry two copies of GGA3, how does GGA3 knockout fit the dystrophies found in AD? In collaboration with Rudolph Tanzi’s lab at Massachusetts General Hospital, Boston, Tesco now reports a GGA3 variant that associates with late-onset AD. Among 966 cases and 427 controls from the NIMH AD Genetics Initiative cohort, Lomoio and colleagues found a rare insertion mutation, Ins545T, in eight people from four AD families; six of those people had late-onset AD. In the AD Neuroimaging Initiative (ADNI) cohort, three carriers of this variant were identified, two of whom had mild cognitive impairment.
Could this variant be pathogenic? When Lomoio and colleagues overexpressed it in hippocampal neurons, it distributed normally throughout the cells, indicating it is normally made and transported. However, in GGA3 knockout neurons, adding the mutant did not rescue BACE1 trafficking or neuron swelling, suggesting the insertion is a loss-of-function mutation (see image below).
No Help from Mutant GGA3. In GGA3 knockout neurons, adding an AD-linked GGA3 mutant did not rescue axon swelling (third panel), indicating it is a loss-of-function mutation. [Courtesy of Lomoio et al., Science Translational Medicine, 2020.]
Edoardo Moretto and Marc Aurel Busche at University College London noted that this mutation is in the hinge region of GGA3, the portion thought to be responsible for summoning clathrin to vesicles. “Whether this GGA3 mutation could directly affect this unconventional sorting of BACE1, or how else it affects BACE1 degradation, remains to be clarified,” they wrote (see full comment below). It also remains to be seen if BACE1 inhibitors can prevent any axonal dystrophies that might be caused by the loss-of-function variant.
If the axonal dystrophies in AD are related to GGA3 loss of function and BAC1 accumulation, then Lichtenthaler thinks tracking that axonal damage may be a way to measure BACE inhibitor effects in early stage disease. “Analyzing axonal damage may provide a straightforward assay to demonstrate that BACE inhibitors not only lower Aβ but also reduce an early form of neurotoxicity,” he said. Scientists are still considering BACE inhibitors for early prevention trials.—Chelsea Weidman Burke
References
News Citations
Research Models Citations
Paper Citations
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007 Jun 7;54(5):721-37. PubMed.
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129-40. PubMed.
Further Reading
Papers
- Santosa C, Rasche S, Barakat A, Bellingham SA, Ho M, Tan J, Hill AF, Masters CL, McLean C, Evin G. Decreased expression of GGA3 Protein in Alzheimer's disease frontal cortex and increased co-distribution of BACE with the amyloid precursor protein. Neurobiol Dis. 2011 Jul;43(1):176-83. PubMed.
Primary Papers
- Lomoio S, Willen R, Kim W, Ho KZ, Robinson EK, Prokopenko D, Kennedy ME, Tanzi RE, Tesco G. Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer's disease trigger BACE1 accumulation in axonal swellings. Sci Transl Med. 2020 Nov 18;12(570) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
German Center for Neurodegenerative Diseases (DZNE)
This is a highly interesting and technically well-controlled study, convincingly demonstrating how GGA3-mediated changes in BACE1 trafficking lead to axonal damage, which also happens early in AD pathogenesis.
This new study is the latest in a series from the Tesco lab on the role of GGA3 in BACE1 trafficking and stability. Collectively, the new study and the previous ones make a strong case for changes in GGA3 abundance or function early in AD pathogenesis. The pathophysiological relevance of the study is further strengthened with the discovery of a new loss-of-function mutation in GGA3 correlating with LOAD.
It will be interesting to see whether more genetic mutations in GGA3 will be found in AD patients and what the mechanisms are that cause the GGA3 loss early in AD pathogenesis. The study also demonstrates that inhibition of BACE1 improves the axonal damage, suggesting that local production of Aβ contributes to that damage.
The analysis of axonal damage may provide a straightforward assay for the clinically used BACE inhibitors to demonstrate that they not only lower Aβ, but also reduce an early form of neurotoxicity, which would be particularly helpful to know for the envisaged primary and secondary prevention studies with BACE inhibitors.
NOVA Medical School
This work increases our understanding of how trafficking regulation is essential for neuronal function, especially regarding GGA3 in neurons. GGAs are clathrin and ARF-binding proteins that typically help sort proteins from the trans-Golgi network to endosomes.
Here, the authors show that GGA3 polarizes to dendrites. I was surprised by the authors' lack of interest in dendrites, where GGA3 is most likely to serve an essential function, given its dendritic predominance. They also looked at axons and found axonal swellings in GGA3 KO neurons. In axons, BACE1 traffics with GGA3 and accumulates in axonal swellings. In vivo, GGA3 KO mice have axonal swelling and 5xFAD mice do worse without GGA3. To link with AD, the authors found a rare variant that the geneticists had missed, an insertion in GGA3, that cannot rescue axonal swellings, indicating that it may be pathological.
If BACE1 is stalled and accumulates in the axonal swellings, I predict that there will be more Aβ in these swellings even if in vivo extracellular Aβ is not changing. The lack of data or discussion on intracellular Aβ accumulation in GGA3 axonal swellings was surprising, given the phenotype recovery upon inhibition of Aβ production.
I was also surprised by the lack of discussion about how GGA3 may be affecting BACE1 transport. Does GGA3 only regulate BACE1 transport? GGA3 also binds to ARF6, which regulates BACE1 endocytosis—is that altered in GGA3 KOs?
One problem in the studies with BACE inhibitors was the lack of non-biased approaches. The experiments focused on Aβ and not on swellings, but what else is going on? BACE1 may have other functions that can be inhibited using BACE inhibitors.
UK DRI at UCL, University College London, UK
University College London
This elegant paper from Lomoio et al. supports the notion that axonal defects, and swelling in particular, are primary events in the pathogenesis of Alzheimer’s Disease (AD), and provides novel mechanistic insights.
The authors build on previous work (Koh et al., 2005; Tesco et al., 2007) and describe the role of the Golgi-localized γ-ear-containing ARF binding protein 3 (GGA3) in BACE1 trafficking and degradation. The two proteins are shown here to undergo axonal transport in the same organelles, possibly lysosomes, at least to some extent. The depletion of GGA3 in neurons was shown to induce axonal swelling by BACE1-positive vesicles.
Interestingly, these organelles did not co-localize with lysosomal markers pointing to a defect in the sorting of BACE1 to the degradation pathway, either by defective maturation of early endosomes to late endosomes, or due to impaired fusion of early endosomes to lysosomes. As synaptophysin-GFP did not accumulate in axonal swellings in GGA3 knockout neurons, this suggests a specific effect of GGA3 depletion on BACE1-containing organelles, which is supported by the previously described direct interaction between BACE1 and GGA3. The absence of an effect of synaptophysin also suggests that the observed swelling was not caused by a general defect in lysosome activity, an otherwise candidate mechanism given the role of GGA3 in sorting of hydrolases (Puertollano and Bonifacino, 2004).
Interestingly, this focal accumulation of BACE1-containing organelles in axonal swellings could conceivably increase APP processing toward Aβ production at these sites. It is widely accepted that the vast majority of APP processing occurs inside endosomes (Haas et al., 2012). As a result, axonal swellings might represent focal sources of Aβ, hypothetically leading to the formation of plaques at these locations, as previously suggested by others (Gowrishankar et al., 2017) but not yet clearly demonstrated. On the other hand, axonal swelling could also lead to an engulfment of axons, with more general downstream impairment of axonal transport.
Axonal transport is crucial for several processes within neurons, from neurotrophin signalling to positioning of mitochondria, and delivery of synaptic vesicles’ precursors to synaptic terminals. Defects in axonal transport are increasingly being associated with several neurological diseases (Sleigh et al., 2019), and have, at least in some cases, been shown to be early pathological events preceding neuronal death in vivo (Smith et al., 2007; Bilsland et al., 2010).
Surprisingly, BACE1 inhibition was found to eliminate the presence of axonal swellings. This is possibly due to a reduction in the levels of BACE1 targeted to degradation, as inhibition might make the protein more stable at the plasma membrane and less likely to need replacement. In contrast, it is possible that BACE1 activity and production of Aβ inside the travelling organelles could affect their own transport and the maturation/fusion to late endosomes/lysosomes. This specific point would need further investigation to be fully elucidated, especially considering the interest in BACE1 inhibition as a potential therapy for early AD.
Another interesting finding of this work is the identification of an Indel in the GGA3 coding gene in a number of patients affected by late-onset AD. This mutation, which was unable to rescue axonal swelling in GGA3 knockout neurons, occurs in the hinge region of GGA3, which is believed to be responsible for clathrin recruitment to vesicles (Bonifacino, 2004). Presence of clathrin on lysosomes/autophagosomes has been previously described and these organelles appear to exchange clathrin-coated vesicles with endosomes (Traub et al., 1996; Rong et al., 2012). Whether this GGA3 mutation could directly affect this unconventional sorting of BACE1, or how else it affects BACE1 degradation, remains to be clarified.
These findings altogether suggest that impaired GGA3-mediated degradation of BACE1, possibly exacerbated by the secretase exaggerated activity, might be involved in the early stages of AD pathogenesis.
A crucial open question that remains is what role tau plays into this picture. As a microtubule stabilizing protein, tau crucially regulates axonal stability and transport. Tau hyperphosphorylation and unbinding from microtubules, as observed in AD, are known to cause axonal instability and axonal transport defects (Zempel and Mandelkow, 2014). Tau could thus synergistically contribute in causing axonal transport defects of BACE1 containing endosomes, thus promoting the formation of Aβ producing swellings (Busche and Hyman, 2020).
References:
Koh YH, von Arnim CA, Hyman BT, Tanzi RE, Tesco G. BACE is degraded via the lysosomal pathway. J Biol Chem. 2005 Sep 16;280(37):32499-504. PubMed.
Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007 Jun 7;54(5):721-37. PubMed.
Puertollano R, Bonifacino JS. Interactions of GGA3 with the ubiquitin sorting machinery. Nat Cell Biol. 2004 Mar;6(3):244-51. PubMed.
Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb Perspect Med. 2012 May;2(5):a006270. PubMed.
Gowrishankar S, Wu Y, Ferguson SM. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J Cell Biol. 2017 Oct 2;216(10):3291-3305. Epub 2017 Aug 7 PubMed.
Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G. Axonal transport and neurological disease. Nat Rev Neurol. 2019 Dec;15(12):691-703. Epub 2019 Sep 26 PubMed.
Smith KD, Kallhoff V, Zheng H, Pautler RG. In vivo axonal transport rates decrease in a mouse model of Alzheimer's disease. Neuroimage. 2007 May 1;35(4):1401-8. PubMed.
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20523-8. Epub 2010 Nov 8 PubMed.
Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004 Jan;5(1):23-32. PubMed.
Traub LM, Bannykh SI, Rodel JE, Aridor M, Balch WE, Kornfeld S. AP-2-containing clathrin coats assemble on mature lysosomes. J Cell Biol. 1996 Dec;135(6 Pt 2):1801-14. PubMed.
Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, Li L, Chen S, Xi J, Yu L. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012 Sep;14(9):924-34. Epub 2012 Aug 12 PubMed.
Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014 Dec;37(12):721-32. Epub 2014 Sep 12 PubMed.
Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer's disease. Nat Neurosci. 2020 Oct;23(10):1183-1193. Epub 2020 Aug 10 PubMed.
Tufts University School of Medicine
Tufts University School of Medicine
We thank our colleagues for taking the time to read our study and provide feedback.
We would like to address some of Dr. Almeida’s valuable comments. Here a few points
1) As stated in the paper, GGA3 localizes to both dendrites and axons both in vivo and in vitro, with a slight preference for dendritic targeting as indicated by its polarity index (1.85). Protein highly polarized in the somatodendritic compartment will display a much higher polarity index (e.g., TfR 4.3 and AP1 10.92). Moreover, under normal conditions BACE1 localizes mostly to presynaptic terminals and accumulates in dystrophic neurites around amyloid-β plaques. These, together with evidence supporting a role for axonal trafficking dysfunction in AD early pathophysiology, have been the reasons that drove us to study GGA3 and BACE1 interaction in the axon.
2) In the discussion paragraph, we explain that our data support previous work, showing that axonal pathology is associated with intraneuronal amyloid-β accumulation and represents an early event in AD. Several studies have reported the presence of intraneuronal amyloid-β in dystrophic neurites in AD brains and in AD mouse models. Similarly, various studies have shown that BACE1 accumulates in dystrophic neurites around senile plaques. Our findings are in line with the previously proposed idea that axonal swellings could form because of impaired axonal transport and promote aberrant amyloid-β generation. If aberrant amyloid-β generation occurs locally at sites of blockage (for instance BACE1 accumulation in Gga3 -/- swellings), then amyloid deposition may occur as a result of focally increased secretion of amyloid-β toxic species or lysis of amyloid-β-enriched axonal swellings.
3) Regarding GGA3 regulation of BACE1 axonal transport, as stated in the manuscript, we strongly believe that according to our trafficking data GGA3 plays a role in transporting BACE1 back to the soma where mature lysosomes reside. We also extensively discuss that our findings indicate that GGA3 loss of function, due to genetic deletion or to an AD-linked GGA3 variant, induces an impairment of BACE1 retrograde trafficking, most likely by affecting the subset of vesicles that co-transport both proteins and are mainly retrograde directed. As a consequence, BACE1 cannot be trafficked back to the soma, where it is normally degraded in mature lysosomes, and starts accumulating in the axon. BACE1 dysfunctional anterograde transport is most likely secondary to increased axonal production of amyloid-β, which, in turn, causes axonal swellings both in vitro and in vivo. Accordingly, β- or γ-secretase pharmacological inhibition rescues BACE1 defective trafficking and axonal buildup in vitro. Moreover, in vivo BACE inhibition prevents hippocampal and cerebellar axonopathy in Gga3 -/- mice.
4) With regard to BACE inhibition experiments, we extensively investigated the effect of the pharmacological treatment on axonal swellings and BACE1 axonal trafficking. We found that BACE inhibitors were able to prevent the swelling phenotype both in vitro and in vivo and restored BACE1 axonal trafficking. We are aware of the fact that BACE1 process a diverse array of substrates besides APP, but so far none of them has been directly correlated with axonopathy. We’ll need more studies in order to understand the effect of BACE inhibition on the many substrates.
Make a Comment
To make a comment you must login or register.