PINK1 Can Act Alone to Destroy Mitochondria, But Parkin Helps
Quick Links
The protein parkin just got a demotion. While it once was considered essential for disposal of worn-out mitochondria, a paper in the August 20 Nature proposes that while parkin does amplify the process of mitophagy, its partner PINK1 is perfectly capable of doing the job all on its own. Both PINK1 and parkin mutations cause familial Parkinson’s and are potential therapeutic targets. Based on the new results, drug discovery research could prioritize PINK1, suggested study co-first author Lesley Kane of the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. Moreover, Kane and colleagues in the laboratory of senior author Richard Youle cemented a previously reported link between mitophagy and two amyotrophic lateral sclerosis genes. They are the kinase TBK1 and its substrate optineurin (OPTN), which recruits the autophagosome to damaged mitochondria.
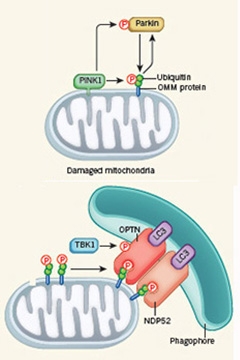
Pathway to Peril.
PINK1 phosphorylates ubiquitinated proteins on the outer mitochondrial membrane. It also phosphorylates parkin, which adds ubiquitin molecules to OMM proteins to create additional PINK1 substrates (top). The phosphorylated ubiquitin chains then recruit the adapter proteins OPTN and NDP52, which in turn summon the autophagy machinery (bottom). [Courtesy of Nature News & Views.]
Improper disposal of old or damaged mitochondria contributes to both ALS and Parkinson’s (see Jul 2015 news; Aug 2013 news). Scientists are investigating how normal mitophagy occurs in order to eventually understand how disease perturbs it.
Researchers initially pictured healthy mitophagy as a linear pathway, wrote Noriyuki Matsuda and Keiji Tanaka of the Tokyo Metropolitan Institute of Medical Science in a commentary accompanying the Nature paper. In this scenario, damaged mitochondria accumulate PINK1, which phosphorylates parkin, which in turn ubiquitinates proteins on the organelle’s outer membrane. The ubiquitin signal brings to the scene proteins called autophagy receptors. These bind autophagy mediators such as LC3, which coat the membranes that gobble the unwanted mitochondria and become the autophagosome (see Feb 2010 news; Kazlauskaite et al., 2014).
However, recently researchers reported that PINK1 phosphorylates not only parkin, but also directly phosphorylates ubiquitin, too, and this action brings parkin to the mitochondrial surface (Shiba-Fukushiima et al., 2014; Okatsu et al., 2015; Koyano et al., 2014; Kane et al., 2014). Last year, scientists also discovered that optineurin acted downstream of parkin, indicating it could be one of the autophagy receptors (see Oct 2014 news). Youle’s lab now adds a loop-the-loop to that linear pathway, which relegates parkin to the role of optional player (see image above right). When it’s there, parkin does ubiquitinate mitochondrial proteins that then act as substrates for PINK1. But it is not necessary. PINK1 can also start mitophagy all by itself. In the latter case, the occasional ubiquitination of mitochondrial proteins, due to natural protein turnover, provides enough substrate for PINK1 to phosphorylate and get mitophagy going.
If something goes wrong with mitophagy, cells fill up with sickened mitochondria. To probe the components of the disposal process, the researchers created a basic cell system. Kane, with co-first authors Danielle Sliter of NINDS and Michael Lazarou, who has since moved to Monash University in Melbourne, Australia, began with HeLa cells. These make PINK1, but not parkin, so the authors added that gene for their experiments. Using gene editing (see Sep 2014 news), they knocked out all five known autophagy receptors, TAX1BP1, NDP52, NBR1, p62, and OPTN, so they could add each one back individually to see how it affected mitophagy. They called these quintuple knockouts pentaKOs.
The authors damaged the mitochondrial respiratory chain in the cells with a combination of oligomycin and antimycin A. In normal cells, this induces mitophagy. As indicators for mitophagy, they measured mitochondrial protein and DNA in the cells, quantifying the inner mitochondrial membrane protein cyclooxygenase II (COXII) by western blotting and the mitochondrial chromosome directly by immunofluorescence for DNA. If COXII and mitochondrial DNA were present, Kane and colleagues concluded that the cell had failed to dispose of the organelle; if those markers were gone, they assumed mitophagy was working. Other researchers have used mitochondrial DNA to assess whether the organelle remains intact (Wong and Holzbaur, 2014), and Youle’s group chose COXII as their protein marker because it resides inside the organelle and is encoded by mitochondrial DNA, Kane said. “It is hard to say that it is absolute proof of mitophagy,” she conceded, adding that is why the authors used two separate measures.

No reception. DAPI stains nuclear DNA (cyan), while antibodies to DNA preferentially bind mitochondrial DNA (green). When mitochondria are poisoned, wild-type cells appropriately digest them and the green disappears. PentaKOs lack autophagy receptors, so the cell cannot get rid of the sick mitochondria and their DNA persists. Cells expressing at least one mitophagy receptor, NDP52 or OPTN, can dispose of mitochondria because the receptors are redundant. Cells missing both those receptors cannot, and the mitochondrial DNA remains. [Courtesy of Lazarou et al., 2015.]
In normal cells, the drug cocktail destroyed much of the COXII and mitochondrial DNA within 24 hours, confirming active mitophagy. Not so in the pentaKOs. When the authors added autophagy receptors back individually, only NDP52 and OPTN, and to a lesser extent TAX1BP1, restored proper mitophagy (see image above). Therefore, these must be the autophagy receptors that are specific for phosphorylated ubiquitin chains on damaged mitochondria. Though the receptors have redundant functions in these cell cultures, they are expressed differentially throughout the body, with OPTN prevalent in neurons, Kane said.
Why were only certain autophagy receptors involved in mitophagy? The authors surmised it might have to do with the unique kind of phosphorylation, on serine 65, that PINK1 adds to ubiquitin molecules (Wauer et al., 2015). It may call out specifically to only these receptors. To test whether ubiquitin phosphorylated at Ser65 would be sufficient to recruit OPTN and NDP52, the researchers created HeLa cells in which a PINK1 construct was inducibly recruited to healthy mitochondria. They left parkin out of this experiment because it would overload the mitochondria with ubiquitin chains, Kane said. Rather, they relied on occasionally ubiquitinated outer-membrane proteins labeled for natural turnover as potential substrates for PINK1 Ser65 phosphorylation. When they dispatched PINK1 to the mitochondria, it did recruit OPTN and NDP52 on its own, whereas a deactivated PINK1 incapable of phosphorylation did not, demonstrating that PINK1 needs to phosphorylate a substrate to do so. To confirm that the substrate is ubiquitin, the authors expressed mutant OPTN and NDP52 missing their ubiquitin-binding domains; these could not find the mitochondria.
This indicated PINK1 phosphorylates ubiquitin and recruits autophagy receptors, all in the absence of parkin. But can the cell complete mitophagy without parkin? To find out, the authors turned to the fluorescent, pH-sensitive mitochondrial marker mKeima, which changes color when the mitochondria land in acidic lysosomes. Sorting cells by color, they observed that 3 to 5 percent of cells executed mitophagy with just OPTN and PINK1. Adding parkin raised the proportion of cells to nearly 24 percent. Thus, PINK1 can work alone, but parkin adds a big boost.
Youle and colleagues suspected that OPTN and NDP52 would work by recruiting LC3, but they were surprised. The mitochondria of double mutants missing both autophagy receptors still collected some LC3, but pentaKOs did not. The authors concluded that other receptors such as p62 might pull in LC3, but not induce mitophagy on their own.
Then what were OPTN and NDP52 needed for? In the NDP52/OPTN double knockouts, recruitment of other parts of the autophagy machinery, e.g., WIPI1 and DFCP1, was subpar. The PINK1 pathway leading to OPTN and NDP52 must somehow recruit these other components of the complete autophagosome, Kane said.
“This thorough study emphasizes an indispensable role for PINK1 in generating phospho-ubiquitin,” commented Helene Plun-Favreau of University College London, who was not involved in the work. “Parkin, on the other hand, seems to amplify the signal … as opposed to being indispensable for mitophagy” (see full comment below).
Dispensable, but still important. “Familial Parkinson’s disease can be caused by mutations in either PINK1 or parkin,” Matsuda and Tanaka wrote in their commentary. “Parkin clearly cooperates with PINK1 and has an important role in PINK1-mediated mitophagy under physiological conditions.”
And what of the ALS link? Since OPTN mutations cause familial ALS (see May 2010 news), the study authors probed how disease-linked mutations might affect mitophagy. ALS-linked OPTN mutants were not recruited to mitochondria and did not support mitophagy. The ALS gene TBK1 phosphorylates OPTN, activating it (see Feb 2015 news; Mar 2015 news). Cells lacking NDP52, which relied only on OPTN for mitophagy, could not destroy mitochondria if TBK1 was also missing. This confirmed that optineurin needs the kinase to degrade mitochondria.
The work suggests that OPTN and TBK1’s role in ALS involves mitophagy, and that mitophagy might be a point of overlap between the degeneration of motor neurons in ALS and that of dopaminergic neurons in Parkinson’s, commented Tim Harris of Biogen in Cambridge, Massachusetts. Harris did not participate in the study. Kane cautioned that more work would be necessary to confirm that the whole PINK1/parkin pathway contributes to ALS pathogenesis in people.
“Mitophagy is an extremely important process as far as neurodegeneration is concerned. We need to understand more about it,” Harris said. Youle’s paper on HeLa cells is a starting point, he said, but the same system should be studied in neurons. Kane responded, “We feel these cells are a good basic tool. We completely agree that future studies need to include not just neurons, but dopaminergic neurons.” She noted that a body of work in neurons and mice already indicates the PINK1/parkin pathway turns on in relevant cells (see Jan 2008 news; Pickrell et al., 2015).—Amber Dance
References
News Citations
- Juvenile Lysosomes and Worn-Out Mitochondria Clog Axons in ALS Model
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- ALS, Parkinson’s Proteins Co-Mingle in Mitochondria Destruction Pathway
- Optineurin Mutations Cause ALS, If Not Glaucoma
- TANK-Binding Kinase 1 Rumbles in as New ALS Gene
- Second Study Salutes TANK-Binding Kinase 1 as ALS Gene
- Pink Fission—Serving Up a Rationale for Parkinson Disease?
Series Citations
Paper Citations
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014 May 15;460(1):127-39. PubMed.
- Shiba-Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Ryu KY, Nukina N, Hattori N, Imai Y. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 2014 Dec;10(12):e1004861. Epub 2014 Dec 4 PubMed.
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015 Apr 13;209(1):111-28. Epub 2015 Apr 6 PubMed.
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 Jun 5;510(7503):162-6. Epub 2014 Jun 4 PubMed.
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014 Apr 28;205(2):143-53. Epub 2014 Apr 21 PubMed.
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014 Oct 21;111(42):E4439-48. Epub 2014 Oct 7 PubMed.
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015 Feb 3;34(3):307-25. Epub 2014 Dec 19 PubMed.
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron. 2015 Jul 15;87(2):371-81. PubMed.
External Citations
Further Reading
Papers
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008 Dec 1;183(5):795-803. Epub 2008 Nov 24 PubMed.
- Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson's disease. Trends Mol Med. 2011 Mar;17(3):158-65. PubMed.
- Wong YC, Holzbaur EL. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy. 2015;11(2):422-4. PubMed.
- Norris KL, Hao R, Chen LF, Lai CH, Kapur M, Shaughnessy PJ, Chou D, Yan J, Taylor JP, Engelender S, West AE, Lim KL, Yao TP. Convergence of Parkin, PINK1, and α-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J Biol Chem. 2015 May 29;290(22):13862-74. Epub 2015 Apr 10 PubMed.
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014 Feb 18;33(4):282-95. Epub 2014 Jan 20 PubMed.
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012 Jun 29; PubMed.
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012 Feb 14;22(2):320-33. PubMed.
Primary Papers
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015 Aug 20;524(7565):309-14. Epub 2015 Aug 12 PubMed.
- Matsuda N, Tanaka K. Cell biology: Tagged tags engage disposal. Nature. 2015 Aug 20;524(7565):294-5. Epub 2015 Aug 12 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
Nearly 10 years ago, the kinase PINK1 and the E3 ubiquitin ligase Parkin, two proteins associated with autosomal-recessive Parkinson’s disease (PD), were identified as key players in the selective degradation of damaged mitochondria (mitophagy). Since then, we have been presented with a central role for both with stabilization of active PINK1 at the mitochondria recruiting Parkin, Parkin redecorating the outer mitochondrial membrane with ubiquitin, ubiquitin recruiting the autophagy and the proteasome machinery, and ultimately inducing mitophagy.
The identification of PINK1 as the first ubiquitin kinase (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014) has considerably improved our understanding of the mitophagy pathway. Now, this thorough study by Richard Youle’s group emphasizes how indispensable PINK1 is because by generating phospho-ubiquitin, the essential ligand for the two primary autophagy receptors (OPTN and NDP52), it recruits the upstream autophagy machinery to mitochondria, and ultimately induces mitophagy.
Parkin, on the other hand, seems to amplify the mitophagy signal by generating more ubiquitin substrate for PINK1 to phosphorylate, as opposed to being indispensable for mitophagy. Whether Parkin is indispensable for mitophagy in HeLa cells or other E3 ubiquitin ligases can play the same role remains to be determined.
Not only does this study clarify the respective roles of PINK1 and Parkin in mitophagy (and show a new PINK1-dependant/Parkin-independent model of autophagy) but it also highlights the probable important role of mitophagy in the pathogenesis of neurodegenerative conditions other than PD. OPTN and DDF52 are associated with primary angle glaucoma, amyotrophic lateral sclerosis, and Crohn’s disease. Why mutations in PINK1, Parkin, OPTN, NDF52, but also other genes such as VCP and MFN2 can all interfere with the mitophagy pathway while leading to different neurodegenerative conditions may be key in understanding selective cellular vulnerability.
References:
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014 Apr 28;205(2):143-53. Epub 2014 Apr 21 PubMed.
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014 May 15;460(1):127-39. PubMed.
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 Jun 5;510(7503):162-6. Epub 2014 Jun 4 PubMed.
McGill University
McGill University
Commentary: This paper presents an exciting surprise to Parkin/PINK1 and mitophagy community in the form of another important discovery from Richard Youle’s laboratory. The Youle group (among others) had previously discovered the PINK1-dependent Parkin activation and recruitment to damaged mitochondria and the involvement of ubiquitin phosphorylated by PINK1 (pUb) in the process (Narendra et al. 2010; Koyano et al. 2014; Kane et al. 2014; Kazlauskaite et al 2014). These discoveries helped uncover the molecular mechanism of Parkin’s activation (Sauvé et al. 2015; Wauer et al. 2015) from its basal autoinhibited state (Trempe et al. 2013). Here, they show that, while Parkin might be required for efficient mitophagy, the process can still occur independently of Parkin, but relies critically on PINK1’s ubiquitin kinase activity.
The general questions that this study aims to answer are: 1) which autophagy receptors are recruited to mitochondria and are critical for PINK1-dependent mitophagy and 2) does pUb generated by PINK1 have roles in mitophagy besides the allosteric activation of Parkin?
The authors generated gene knockouts of five autophagy cargo receptors to impair mitophagy and show that the reintroduction of only NDP52 or Optineurin (OPTN) rescued the impairment. This rescue occurs even in the absence of Parkin. NDP52 and OPTN (and its phosphorylation by TBK1) have been investigated previously for their roles for xenophagy (Thurston et al. 2009; Wilds et al. 2011) but their involvement in mitophagy is novel. The absence of Parkin helped the authors discriminate better the autophagy receptors that are critical and those that are dispensable. Previous studies (Geisler et al. 2010) had suggested the recruitment of p62 to mitochondria following Parkin activation, but here it is shown not to be essential for mitophagy. The new study demonstrates the value of COXII and mtDNA depletion as markers for mitophagy as MFN1 (a common marker for Parkin’s activity and mitophagy) and TOM20 can be targeted to the proteasome directly by Parkin-mediated ubiquitination in the absence of autophagy.
Youle’s group also discovers another role for pUb: the recruitment of NDP52 and OPTN. They demonstrate that NDP52 and OPTN are preferentially pulled down by phosphomimetic ubiquitin from HeLa cells. The cargo receptor p62 doesn’t have this property. This part of the manuscript will require some additional proof. For example, some experiments were done using phosphomimetic ubiquitin, which doesn’t capture all the functional characteristics of pUb (Ordureau et al. 2015). Also, the use of a Parkin overexpression system in the latter experiments puts the assertion about the Parkin-independence of the process on a difficult footing. Nonetheless, the observations are very significant. Future studies including a paper this week from the Harper group (Heo et al. 2015) will clarify the molecular basis of NDP52 and OPTN recruitment.
Finally, it has been shown that in the absence of NDP52 and OPTN or PINK1 both upstream (ULK1 complex) and downstream components (LC3B) of the autophagy machinery are not effectively recruited, explaining the impairment in mitophagy. The study also shows that mutations in NDP52 and OPTN associated with various diseases including amyotrophic lateral sclerosis (ALS or Lou Gehrig's disease) and Crohn’s disease are impaired in mitophagy and hence indicates that impaired mitophagy might have a contribution towards the observed pathologies. This new study adds significantly to our understanding of mitophagy by assigning roles to different components of the mitochondrial quality control pathway, both upstream and downstream in the process. The findings should also encourage the investigation and development of therapeutics targeting PINK1 activation for the treatment of Parkinson disease.
References:
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010 Feb;12(2):119-31. Epub 2010 Jan 24 PubMed.
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015 Oct 1;60(1):7-20. Epub 2015 Sep 10 PubMed.
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014 Apr 28;205(2):143-53. Epub 2014 Apr 21 PubMed.
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014 May 15;460(1):127-39. PubMed.
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 Jun 5;510(7503):162-6. Epub 2014 Jun 4 PubMed.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008 Dec 1;183(5):795-803. Epub 2008 Nov 24 PubMed.
Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A. 2015 May 26;112(21):6637-42. Epub 2015 May 12 PubMed.
Sauvé V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, Gehring K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015 Aug 7; PubMed.
Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009 Nov;10(11):1215-21. Epub 2009 Oct 11 PubMed.
Trempe JF, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013 Jun 21;340(6139):1451-5. Epub 2013 May 9 PubMed.
Wauer T, Simicek M, Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature. 2015 Aug 20;524(7565):370-4. Epub 2015 Jul 10 PubMed.
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011 Jul 8;333(6039):228-33. Epub 2011 May 26 PubMed.
Make a Comment
To make a comment you must login or register.