C9ORF72 Dipeptide Repeat Not Enough for Motor Neuron Disease
Quick Links
Scientists trying to understand what is toxic about the C9ORF72 hexanucleotide expansion, linked to amyotrophic lateral sclerosis and frontotemporal dementia, have implicated two different RNA repeats and five different dipeptide-repeat proteins as potential culprits. In the March 21 Nature Neuroscience online, researchers focus on one of the peptide repeats, poly-glycine-alanine. When expressed in mouse brain, poly(GA) interfered with protein degradation and nucleocytoplasmic transport, report senior author Leonard Petrucelli and colleagues at the Mayo Clinic in Jacksonville, Florida. Poly(GA) aggregates sequestered HR23 proteins, which transport nuclear proteins to the proteasome for degradation. However, the mice were notably free of a major feature of typical C9ORF72 pathology, i.e., the TDP-43 inclusions. The results imply that while poly(GA) clearly contributes to ALS and FTD, it must work with other players to create the full spectrum of neuropathology.

Dipeptide Inclusions:
Green fluorescent protein-poly(GA) chimeras (top), but not GFP controls (bottom), form cytoplasmic inclusions. (Nuclei stained blue.) [Courtesy of Zhang et al., Nature.]
The GGGGCC expansions in the C9ORF72 gene are transcribed in both sense- and antisense directions. Messenger RNAs are then translated into five different polypeptides (two of the six possible reading frames both encode poly-glycine-proline). Some studies suggest the RNAs cause neurodegeneration (see Feb 2016 news; May 2015 news; Oct 2013 news). Others point an accusing finger at the repeat dipeptides, particularly the poly-glycine-arginine and poly-proline-arginine versions. Perhaps that is because arginines target the peptides to the nucleolus, where they prevent normal RNA production (see Aug 2014 news; news; Dec 2014 news). And yet some studies find that poly(GA), not its arginine-rich counterparts, is among the most abundant repeat dipeptides in the brains of C9ORF72 expansion carriers (see Feb 2013 news; Mackenzie et al., 2015).
Petrucelli’s group and others have found that poly(GA) makes neurotoxic inclusions in cultured neurons. It co-aggregates with ubiquitin and p62, disrupting proteasome activity (Yamakawa et al., 2015; Zhang et al., 2014). In the current study, first author Yong-Jie Zhang and colleagues tested whether it would also be neurotoxic in vivo.

Lost Neurons:
Compared to controls (left), mice expressing poly(GA) had fewer neurons in the motor cortex (top) and hippocampus (bottom). [Courtesy of Zhang et al., Nature.]
The authors designed a transgene that encodes poly(GA) without the repetitive RNA sequences. They packaged it in adeno-associated virus and injected it into the brains of newborn mouse pups, then analyzed them six months later. As a control, the authors made an interrupted poly(GA) gene, with a proline inserted after every fifth GA repeat. Both constructs were expressed at similar levels in the mice.
In the neurons of the cortex and many other regions of the animals’ brains, poly(GA) formed cytoplasmic, and occasionally nuclear, inclusions. Most of these contained ubiquitin as well. The interrupted poly(GA) construct did not aggregate.
Only aggregation-capable poly(GA) was neurotoxic. The brains of mice expressing poly(GA) weighed about one-third less than those that received the interrupted gene or a control construct with only green fluorescent protein (GFP). The poly(GA) mouse brains contained fewer neurons in the cortex, hippocampus, and cerebellum (see image above). Poly(GA) mice also had behavioral abnormalities. They fell off a rotating rod sooner than controls, indicating motor neuron defects, and had trouble learning to associate white noise with a foot shock.
Petrucelli said he is not sure if the large poly(GA) aggregates are the toxic species, or if smaller species such as oligomers do the real damage. Whatever their size, how did poly(GA) peptides cause neurodegeneration? The authors knew from the cell-culture studies that it interfered with the proteasome but were unsure how. In a previous study, scientists had found that poly(GA) bound to HR23A and HR23B, which ferry ubiquitinated proteins from the nucleus to the proteasome (May et al., 2014). Zhang and colleagues suspected that poly(GA) might interfere with this process.
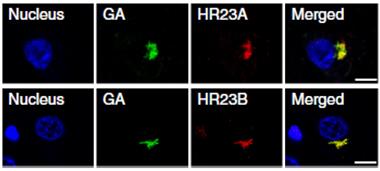
Double trouble:
Poly(GA) peptides aggregate with HR23 proteins in the cytoplasm of neurons from people who died of C9ORF72-based disease. [Courtesy of Zhang et al., Nature.]
While HR23 proteins are normally diffuse in the nucleus, in the brains of poly(GA) mice they aggregated in the cytoplasm and nuclei. Moreover, these aggregates included poly(GA), suggesting the repeat dipeptide sequestered the nuclear HR23 proteins. HR23 proteins also co-aggregated with poly(GA) in hippocampal sections from people who died of FTD or ALS due to the C9ORF72 expansion (see image at left).
This raised the question of how the nuclear HR23 proteins mislocalize to the cytoplasm. Other researchers have reported that cytoplasmic aggregates interfere with nucleocytoplasmic transport (see Dec 2015 news) and C9ORF72 expansions, in particular, seem to block traffic between the two cellular compartments (see Aug 2015 news). Nucleocytoplasmic transport also appeared defective in the poly(GA) mice; both the traffic regulator RanGAP1 and the nuclear pore component Pom121 wound up in poly(GA) inclusions. In addition to recruiting HR23s directly into aggregates, poly(GA) may cause some nuclear pore defect that contributes to HR23 mislocalization, suggested the authors.
To check whether poly(GA) interfered with HR23 function, the authors examined the DNA damage repair protein XPC (xeroderma pigmentosum C). Normally, HR23 proteins stabilize XPC and protect it from degradation. In the poly(GA) mice, XPC levels were half that of controls, indicating HR23 was not working.
If loss of HR23 function caused the toxicity in the mouse brains, Zhang and colleagues reasoned, then adding more HR23 might rescue the neurons. Because HR23B can compensate for lack of either HR23A or B, Zhang expressed it in primary neurons along with poly(GA). This returned XPC levels to normal and decreased caspase-3 activation, which was associated with neurotoxicity in the poly(GA) neurons.
Extra HR23B also reduced the aggregation of poly(GA). Though the scientists are unsure how, Petrucelli speculated it might assist the beleaguered proteasome system, preventing aggregation or dissolving the inclusions.
This paper confirms HR23 proteins as players in C9ORF72-based disease, commented Brian Freibaum of St. Jude Children’s Research Hospital in Memphis, Tennessee, who did not participate in the work. HR23A and B also aggregate in neurons of people with Huntington’s disease, spinocerebellar ataxia, and fragile X-associated tremor/ataxia syndrome (Bergink et al., 2006).
Despite the neurodegeneration in the poly(GA) mice, they conspicuously lacked one major feature of C9ORF72-based disease, that is, cytoplasmic TDP-43 inclusions. Other C9ORF72 dipeptides might precipitate those inclusions, Petrucelli suggested. “It will be of great interest to determine if that is true,” said Adrian Isaacs of University College London, who was not involved in the paper (see full comment below). Petrucelli is working on other dipeptide repeat mice, with poly(PR) next in line, he said.
“The confirmation of poly(GA) toxicity in mice is an important step forward and will no doubt further fuel the current debate around the toxic species in C9FTD/ALS,” Isaacs said. “While these new data show poly(GA) can be toxic, they do not clarify which species is ultimately the toxic one in patients,” he cautioned. —Amber Dance
References
News Citations
- Repeat RNAs Hitchhike to the Ends of Neurons, Attacking Neurites
- Antisense RNA from C9ORF72 Repeats Is Likely Culprit in Patient Neurons
- RNA Deposits Confer Toxicity in C9ORF72 ALS
- C9ORF72 Killer Dipeptides Clog the Nucleolus
- C9ORF72’s Dirty Work Done by Problem Proteins
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- RNA Twist: C9ORF72 Intron Expansion Makes Aggregating Protein
- Too Much of a Bad Thing: Protein Aggregates Snarl Nuclear Traffic
- ALS Gene Repeats Obstruct Traffic Between Nucleus and Cytoplasm
Paper Citations
- Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, Neumann M. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015 Dec;130(6):845-61. Epub 2015 Sep 15 PubMed.
- Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, Suzuki N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet. 2015 Mar 15;24(6):1630-45. Epub 2014 Nov 14 PubMed.
- Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, Chew J, Belzil VV, Prudencio M, Stankowski JN, Castanedes-Casey M, Whitelaw E, Ash PE, DeTure M, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014 Oct;128(4):505-24. Epub 2014 Aug 31 PubMed.
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf-Strathmann J, Mann M, Meissner F, Edbauer D. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014 Oct;128(4):485-503. Epub 2014 Aug 14 PubMed.
- Bergink S, Severijnen LA, Wijgers N, Sugasawa K, Yousaf H, Kros JM, van Swieten J, Oostra BA, Hoeijmakers JH, Vermeulen W, Willemsen R. The DNA repair-ubiquitin-associated HR23 proteins are constituents of neuronal inclusions in specific neurodegenerative disorders without hampering DNA repair. Neurobiol Dis. 2006 Sep;23(3):708-16. PubMed.
Further Reading
Papers
- Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 2015 Oct;130(4):525-35. Epub 2015 Jun 2 PubMed.
- Dodd DW, Tomchick DR, Corey DR, Gagnon KT. Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches. Biochemistry. 2016 Mar 8;55(9):1283-6. Epub 2016 Feb 22 PubMed.
- Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016 May 1;25(9):1803-13. Epub 2016 Feb 29 PubMed.
- Edbauer D, Haass C. An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr Opin Neurobiol. 2016 Feb;36:99-106. Epub 2015 Nov 8 PubMed.
Primary Papers
- Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, Lin WL, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD, Petrucelli L. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016 May;19(5):668-77. Epub 2016 Mar 21 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
Zhang et al. convincingly show that in mice, overexpression of polyGA causes neurodegeneration, which is dependent on the ability of polyGA to aggregate. The confirmation of polyGA toxicity in mice is an important step forward and will no doubt further fuel the current debate around the toxic species in C9ORF72-based FTD/ALS. Our work showed that polyGA is toxic to adult Drosophila neurons, but that it is much less potent than polyGR and polyPR (Mizielinska et al., 2014). This indicates that the levels of these proteins in patients will be an important factor. Therefore, while these new data show that polyGA can be toxic, they do not clarify which species is ultimately the toxic one in patients. That will require a better understanding of not only the levels of each species in patients but also their conformation; indeed, the authors here suggest polyGA oligomers may be the culprit.
A powerful aspect of the study is the ability to compare polyGA toxicity to the Petrucelli lab’s pure repeat mouse model that utilizes the same AAV system but generates polyGA, polyGP, polyGR, and RNA foci (Chew et al., 2015). Importantly, TDP-43 pathology is much greater in the pure repeat mice, indicating that polyGA is not solely responsible for TDP-43 aggregation. It will be of great interest to determine whether expression of the other individual dipeptide repeat proteins (DPRs), such as polyGR and polyPR, lead to TDP-43 pathology. An intriguing possibility is that more than one DPR may be required for TDP-43 aggregation. It is also possible the RNA repeats rather than DPRs drive TDP-43 pathology.
Another intriguing aspect of the work is the identification of nuclear pore defects in polyGA mice. This is because work in Drosophila and yeast indicate that either polyGR/PR or repeat RNA are responsible for the nucleocytoplasmic defects identified in these model systems (Freibaum et al., 2015; Zhang et al., 2015; Jovicic et al., 2015; Boeynaems et al., 2016). Further investigation of the specific effects of each species on nucleocytoplasmic transport should give new insight into this emerging pathway and its role in C9FTD/ALS.
References:
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EM, Partridge L, Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014 Sep 5;345(6201):1192-1194. Epub 2014 Aug 7 PubMed.
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015 Jun 5;348(6239):1151-4. Epub 2015 May 14 PubMed.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015 Sep 3;525(7567):129-33. Epub 2015 Aug 26 PubMed.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015 Sep 3;525(7567):56-61. Epub 2015 Aug 26 PubMed.
Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015 Sep;18(9):1226-9. PubMed.
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016 Feb 12;6:20877. PubMed.
Max Planck Institute of Biochemistry
This is an interesting study that adds further important insights to a series of findings that all point toward transport across the nuclear membrane as a process that is especially vulnerable not only in cells that encounter aggregation-prone proteins (Freibaum et al., 2015; Jovicic et al., 2015; Woerner et al., 2016; Zhang et al., 2015), but also in in aging neurons (Mertens et al., 2015).
In this manuscript, the authors describe how expression of poly(GA) repeat-containing proteins leads to the mislocalization and aggregation of RAD23. The appearance of large protein aggregates in the “wrong” cellular compartment is a phenomenon that is also observed in other neurodegenerative diseases. Interestingly, overexpression of RAD23 is sufficient to prevent poly(GA)-induced toxicity in this system, most likely by restoring original RAD23 levels in the nucleus.
It is important to note that impairment of nucleocytoplasmic transport by protein aggregates is not sufficient to explain all the effects of toxicity observed in c9FTD/ALS (or in other neurodegenerative diseases), however, I believe that a better understanding of the factors that maintain the nucleocytoplasmic compartmentalization is necessary to understand many of these diseases, and that transport across the nuclear envelope is a process that warrants further research.
For me, an important open question in this regard concerns the order of events: Can defects in nucleocytoplasmic transport be causative for neurodegeneration, or is the collapse of nucleocytoplasmic compartmentalization an event that happens downstream of another insult?
References:
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015 Sep 3;525(7567):129-33. Epub 2015 Aug 26 PubMed.
Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015 Sep;18(9):1226-9. PubMed.
Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016 Jan 8;351(6269):173-6. Epub 2015 Dec 3 PubMed.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015 Sep 3;525(7567):56-61. Epub 2015 Aug 26 PubMed.
Mertens J, Paquola AC, Ku M, Hatch E, Böhnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, Gonçalves JT, Toda T, Kim Y, Winkler J, Yao J, Hetzer MW, Gage FH. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 2015 Oct 6; PubMed.
Make a Comment
To make a comment you must login or register.