Brain Inflammation Drastically Changes Behavior and Lifespan in Animals
Quick Links
Inflammation is a fickle friend indeed. Some pathways have benefits, while others do great harm. Two new papers add data to the latter category. Scientists led by Li Gan, University of California, San Francisco, report that inflammatory signaling in microglia, brought on when progranulin goes missing, causes mice to groom themselves incessantly. Appearing in the April 24 Early Edition from the Proceedings of the National Academy of Sciences USA, the mouse data show similar compulsive behavior as seen in people carrying progranulin mutations that cause frontotemporal dementia (FTD). The results suggest that hyperactivation via the microglial NFκB-tumor necrosis factor alpha axis may mediate some of the behavioral problems in FTD. A second study in the April 24 Cell Reports links excess immune signaling to premature neurodegeneration and early death in flies. Petros Ligoxygakis, University of Oxford, U.K., reports that knocking back the NFκB signaling pathway extends lifespan, while revving it up shortens it. In both studies, scientists find that dialing back similar inflammatory signals rescues behavior and early death.
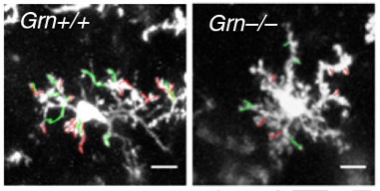
Feeling Around.
Two-photon microscopy in living mice shows that microglia in progranulin knockouts (right) are less vigilant than those from controls (left), poking fewer finger-like extensions (green) and retractions (red) into the environment. [Image courtesy of Krabbe, PNAS.]
People with mutations in the progranulin (PGRN) gene develop FTD, which is marked by compulsive behaviors, social withdrawal, and loss of empathy. PGRN knockout mice recapitulate some of those behaviors, including a tendency to compulsively scratch at their mouths and backs to clean themselves (see Lui et al., 2016). While wild-type mice prefer the company of other mice, PGRN knockouts are just as likely to hang out with inanimate objects (Yin et al., 2010).
These progranulin-deficient mice reportedly express more proinflammatory cytokines, such as TNFα, and have a larger number of active microglia (Yin et al., 2010). Gan and colleagues wondered whether the excess inflammation in these mice might explain their behavior.
To find out, co-first authors Grietje Krabbe, Sakura Minami, and Jon Etchegaray compared PGRN knockout mice with those that also lacked one or both copies of TNFα. While the PGRN knockouts cleaned themselves incessantly, the double transgenics did so about a third less, comparable to wild-type mice. They still spent the same amount of time with another mouse as with an inanimate object, however, suggesting TNFα signaling did not account for the antisocial behaviors.
Compulsive behavior has been traced back to hyperactive medium spiny neurons in the nucleus accumbens (Ahmari et al., 2013). In the present study, whole cell patch-clamp recording confirmed that knocking out PGRN in mice caused these medium spiny neurons to fire faster than normal. However, taking out one or both copies of TNFα slowed that firing back down to wild-type levels.
Microglia seemed especially sensitive to these genetic alterations. In progranulin knockouts, microglia extended fewer finger-like projections into the environment to probe for signs of trouble than controls (see image above). They also reacted more sluggishly to a laser-induced injury (see video below). In fact, knocking out progranulin in microglia alone was enough to elicit the compulsive self-grooming behavior in mice. What’s more, deleting NF-κB—a master regulator of TNFα (see image below)—in the microglia of mice with no progranulin restored this behavior to normal, along with other behaviors that had gone haywire in these mice, including social deficits and excessive marble burying. This suggests NF-κB signaling has a broader influence on behavior.
Together, the results support microglial NF-κB and TNFα signaling as an operative pathway in PGRN-deficient FTD, wrote the authors. “We now have direct proof that microglia abnormalities contribute to FTD-like behavior,” said Gan. “That had not been shown before.” Her group saw similar compulsive behaviors in people with PGRN mutations, such as obsessive hygiene and collecting objects. “They carry out these repetitive behaviors because their circuit drives them to do it—they just can’t stop,” Gan went on. Previous studies confirmed high levels of TNFα in the plasma of these patients (Miller et al., 2013). Gan’s research now suggests these behaviors could be calmed by modulating the inflammatory pathways in microglia. “It provides a new angle for intervention in FTD,” Gan said. She next plans to work out how TNFα changes the intrinsic excitability of medial spiny neurons, and whether it can be reversed with therapy.
“Taken together with other published literature, it is clear that genetic ablation of progranulin leads to profound neuroinflammation and alterations in microglia activities,” wrote Malú Tansey and Thomas Kukar, Emory University School of Medicine, Atlanta, to Alzforum (see full comment below). They cautioned that TNF-deficient mice also lack many other cytokines and chemokines, so drugs that target TNFα would help single out its role.
In the paper by Ligoxygakis and colleagues, first authors Ilias Kounatidis and Stanislava Chtarbanova note that as people age, immune signaling ramps up and inflammation becomes chronic. To find out if that leads to neurodegeneration, the researchers created multiple fly models with mutated negative regulators of the NF-κB immune pathway. By removing the brakes, the authors escalated this signaling. In these fly mutants, neurodegeneration accelerated greatly, as measured by trouble climbing up inside the vials in which they were kept. Whereas wild-type flies lived about 65 days, these only made it to about 25.
Conversely, if the authors genetically knocked down NF-κB signaling, the flies lived an average of 105 days. They scrambled up their vials late into old age, meaning their “healthspan” was also extended. Curiously, while age-related neurodegeneration progressed as usual in these flies, they released twice as much of the adipokinetic hormone (Akh). This caused energy stores to break down into usable sugars and triglycerides, and may explain why the flies were more active, said Ligoxygakis. A similar change in hormone signaling and lifespan extension was reported in mice with reduced NF-κB signaling (Zhang et al., 2013).
“The paper shows for the first time that both increase and decrease of NF-κB activity, and therefore of immune activity, can determine lifespan,” said Ligoxygakis. “It’s all centered on one molecule.” NF-κB could become a therapeutic target for neurodegenerative diseases, or even prolong healthspan, he said, but the challenge will be to modulate this signaling locally in the brain.
“This is a fascinating report using fly models to examine the intersection of aging, neurodegeneration, and metabolism,” wrote Terrence Town, University of Southern California, Los Angeles to Alzforum (see full comment below). “This result hints at an evolutionary basis for control of a glial phenotype that maintains homeostasis and repair.”
“This work is impressive, as it clearly demonstrates that neural NF-κB is a crucial determinant of aging and lifespan in drosophila,” wrote Dongsheng Cai, Albert Einstein College of Medicine, New York, to Alzforum (see full comment below). Together with the previous research in mice, “the case is growing stronger that the hypothalamic NF-κB pathway has a programmatic role in aging and longevity, even though the responsible cell types in the brain, especially in the neuroendocrine region, are still unclear,” he added.—Gwyneth Dickey Zakaib
Feel the burn? Microglia rush to the site of a laser-induced injury in normal mice with intact progranulin (first clip). However, when progranulin is missing, the microglia are more sluggish in their approach (second clip). [Image courtesy of Krabbe, PNAS.]
Media
References
Paper Citations
- Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam MM, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RV Jr, Paz JT, Barres BA, Huang EJ. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016 May 5;165(4):921-35. Epub 2016 Apr 21 PubMed.
- Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, Ding A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010 Dec;24(12):4639-47. PubMed.
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010 Jan 18;207(1):117-28. PubMed.
- Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, Gordon JA, Hen R. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013 Jun 7;340(6137):1234-9. PubMed.
- Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, Criswell LA, Jaeger PA, Stan T, Heggeli KA, Hsu SC, Karydas A, Khan BK, Grinberg LT, Gorno-Tempini ML, Boxer AL, Rosen HJ, Kramer JH, Coppola G, Geschwind DH, Rademakers R, Seeley WW, Wyss-Coray T, Miller BL. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. 2013 Mar 30; PubMed.
- Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013 May 1; PubMed.
Further Reading
Primary Papers
- Krabbe G, Minami SS, Etchegaray JI, Taneja P, Djukic B, Davalos D, Le D, Lo I, Zhan L, Reichert MC, Sayed F, Merlini M, Ward ME, Perry DC, Lee SE, Sias A, Parkhurst CN, Gan WB, Akassoglou K, Miller BL, Farese RV Jr, Gan L. Microglial NFκB-TNFα hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci U S A. 2017 May 9;114(19):5029-5034. Epub 2017 Apr 24 PubMed.
- Kounatidis I, Chtarbanova S, Cao Y, Hayne M, Jayanth D, Ganetzky B, Ligoxygakis P. NF-κB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell Rep. 2017 Apr 25;19(4):836-848. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Southern California
This is a fascinating report using fly models to examine the intersection of aging, neurodegeneration, and metabolism. While flies don’t have a true microglial analog, they do have a multifunctional type of glial cell. Studies from Marc Freeman’s lab demonstrate “phagocytic” or “ensheathing” glia that take on some of the functionality of microglia in mammals. In this paper, ablation of NF-κB signaling impacts the cerebral innate immune/inflammatory response. When the authors dampen NF-κB, there is a shift in glial metabolism that promotes tissue maintenance and repair.
What’s even more exciting is that the Rel (NF-κB analog) pathway signals through and activates a TGF-β-like analog, promoting cell survival. This result hints at an evolutionary basis for control of a glial phenotype that maintains homeostasis and repair. While the authors don’t discuss whether peripheral immune cells infiltrate the brain, one nonetheless wonders whether this occurs in their fly models and ultimately impacts neurodegeneration. …More
The University of Florida College of Medicine
Emory University
Mutations in the GRN gene are a common cause of frontotemporal dementia and lead to loss of progranulin (PGRN) protein function, yet the mechanisms leading to neurodegeneration are still unclear. Complete loss of PGRN in mice and humans leads to lysosome dysfunction, a lysosome storage phenotype, which is accompanied by robust neuroinflammation including astrogliosis and microgliosis. Converging evidence from multiple groups suggest that both lysosome dysfunction and inflammation are important components of disease pathogenesis in FTD caused by GRN mutations. Dissecting which cell types in the brain are dysfunctional and/or driving dysfunction is an important remaining question in the field.
Krabbe et al. performed an elegant set of in vivo and imaging studies to demonstrate OCD-like behavioral alterations (i.e., over-grooming and sociability deficits) in mice lacking progranulin (PGRN) as well as impaired microglia/myeloid cell responses to injury, and medium spiny neuron (MSN) hyperexcitability. To identify the cell type in which Grn deletion mattered, the authors ablated Grn in microglia/myeloid cells and observed increased grooming in mice; conversely, they observed modest reduction in grooming in mice where the master regulator of inflammatory responses (NFκB) was inactivated.…More
Next, to interrogate the role of tumor necrosis factor, given that TNF has been implicated in the pathophysiology of FTD and nearly every neurological disorder, the authors crossed Grn KO mice with TNF KO mice. They found that this mitigated the self-grooming modestly and the associated hyperexcitability of MSNs, but not the sociability deficits. Studies from Stellwagen and Malenka almost 10 years ago revealed that glial-derived TNF plays a key role in homeostatic synaptic scaling, a form of synaptic plasticity (Stellwagen and Malenka, 2016). Therefore, excess production of TNF and many other inflammatory mediators may have contributed to disruptions in this form of plasticity and hyperexcitability.
Overall, Krabbe et al.’s findings reveal a highly dysregulated phenotype of myeloid (microglia) cells in the brain resulting from global ablation of GRN expression. These findings are consistent with those by the recent study by Lui et al., 2016, in which global ablation of Grn was associated with dysregulated microglia/myeloid cells that overproduce inflammatory mediators and over-prune synapses in the thalamus.
Taken together with other published literature, it is clear that genetic ablation of GRN leads to profound neuroinflammation and alterations in microglia activities. Therefore, it is consistent with how the levels of multiple inflammatory cytokines are altered, which is accompanied by decreased phagocytosis and wound-healing responses.
While the genetic cross between GRN KO mice and TNF KO mice is an easy way to interrogate the role of TNF and downstream targets in mediating the hyper-inflammatory phenotype, one limitation of this experiment is that the rescue effect may not be the direct result of TNF inhibition alone. Specifically, TNF-deficient mice also lack many other cytokines and chemokines (including IL-6, IL1β, IL-10, IL-12, and CXCL1) (Harms et al., 2012), and the double KO mouse is likely to be missing those, too. As noted by the authors in the conclusions, pharmacological inhibition of TNF or other cytokines would be a more direct way to ascertain its direct role and certainly merits investigation as a potential therapeutic path forward even if PGRN does not directly bind or negatively regulate TNF receptors, as has been reported by three separate and independent laboratories on TNF-TNFR signaling (Chen et al., 2013; Etemadi et al., 2013; Hu et al., 2014).
Taken together, this work provides additional evidence that loss of PGRN leads to dysregulation of the lysosome-inflammation axis, paving the way for novel therapeutic strategies for FTD.
References:
Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006 Apr 20;440(7087):1054-9. PubMed.
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam MM, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RV Jr, Paz JT, Barres BA, Huang EJ. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016 May 5;165(4):921-35. Epub 2016 Apr 21 PubMed.
Harms AS, Lee JK, Nguyen TA, Chang J, Ruhn KM, Treviño I, Tansey MG. Regulation of microglia effector functions by tumor necrosis factor signaling. Glia. 2012 Feb;60(2):189-202. Epub 2011 Oct 11 PubMed.
Chen X, Chang J, Deng Q, Xu J, Nguyen TA, Martens LH, Cenik B, Taylor G, Hudson KF, Chung J, Yu K, Yu P, Herz J, Farese RV, Kukar T, Tansey MG. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J Neurosci. 2013 May 22;33(21):9202-13. PubMed.
Etemadi N, Webb A, Bankovacki A, Silke J, Nachbur U. Progranulin does not inhibit TNF and lymphotoxin-α signalling through TNF receptor 1. Immunol Cell Biol. 2013 Nov-Dec;91(10):661-4. Epub 2013 Oct 8 PubMed.
Hu Y, Xiao H, Shi T, Oppenheim JJ, Chen X. Progranulin promotes tumour necrosis factor-induced proliferation of suppressive mouse CD4⁺ Foxp3⁺ regulatory T cells. Immunology. 2014 Jun;142(2):193-201. PubMed.
This work is impressive, as it clearly demonstrates that neural NF-κB is a crucial determinant of aging and lifespan in drosophila. The authors’ results are consistent with, and also complementary to, the findings reported from mouse models a few years ago (Zhang et al., 2013). Together with the emergence of many other supportive data for this notion, the case is now strong that the hypothalamic NF-κB pathway has a programmatic role in aging and longevity, even though the responsible types of cells in the brain and especially in the neuroendocrine region of the brain still remain unclear.
References:
Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013 May 1; PubMed. …More
SENS Research Foundation
By coincidence, Richard Miller and colleagues with the NIA's Interventions Testing Program have just published evidence that interventions that extend natural lifespan in rodents also selectively reduce brain inflammation mediated by TNF-α.
References:
Sadagurski M, Cady G, Miller RA. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell. 2017 May 20; PubMed.
Make a Comment
To make a comment you must login or register.