The THIK and Thin of Microglial Surveillance
Quick Links
Long given short shrift by neuroscientists, microglia are blossoming into darling research subjects. The newfound attention comes courtesy of genetic evidence implicating microglia in neurodegenerative diseases and cellular evidence for a role in pruning synapses. While science still knows little about what makes these immune cells tick, that’s beginning to change. At “Microglia in the Brain,” a joint symposium held June 12-16 together with “Common Mechanisms of Neurodegeneration,” in Keystone, Colorado, researchers revealed a potassium channel (THIK-1) that drives microglia to survey their milieu.
In the healthy brain, microglia constantly extend and retract finger-like projections, presumably to feel around for signs of trouble such as foreign invaders or dying neurons, and to sense the functional state of synapses. Repeated scanning of microglia in the brain show that they can probe the entire organ within a few hours. What drives this behavior? Scientists at Keystone agreed that answering this question is fundamental to understanding how microglia function in the brain. Taking a stab at the problem, David Attwell, University College London, reported that an outwardly rectifying potassium channel drives microglial surveillance.

Feeling About.
Images of a mouse hippocampal microglia captured at a single time point (left), and cumulatively over 20 minutes (right), show how it surveys its environment. [Image courtesy of David Attwell and Christian Madry.]
Attwell and collaborators identified the potassium channel by studying cell motility in mouse brain tissue slices. They began by probing responses in the classic laser-injury model, whereby microglia flood in to repair a hole burned by a laser. In this paradigm, two things happen in microglia, he said. Cytosolic calcium levels rise, and potassium exits the cells via an ion channel. However, the responses are independent of each other, said Attwell. He knows this because thapsigargin, which blocks the mobilization of calcium from internal endoplasmic reticulum stores, has no effect on the potassium efflux.
Adding ATP to microglia evokes both these responses, hence they appear to be initiated by purinergic receptor signaling. Microglia abundantly express several of these receptors, which are activated by ATP released by dead and dying cells. Scientists believe that ATP and other purines spur microglia into action to tidy up cellular debris. Attwell showed the P2Y12 purinergic receptors primarily trigger the potassium current. PSB 0739, a P2Y12R antagonist, suppressed it, while blocking other purinergic receptors had no effect on potassium efflux. “This is phenomenally important work,” noted Richard Ransohoff from Biogen in Cambridge, Massachusetts. “It gives us a molecular explanation [for motility] and therefore numerous genetic and pharmacological tools to manipulate it,” he told Alzforum.
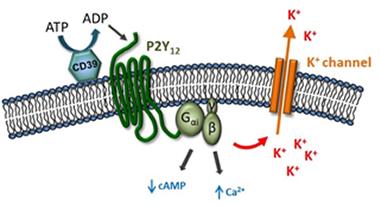
Channeling Ions.
Purinergic receptors may trigger release of potassium in microglia. [Image courtesy of David Attwell.]
While the P2Y12 receptor may trigger the potassium release, which ion channel conducts it? Attwell found that blocking voltage-dependent K+ channels had no effect. This left inward rectifying and two-pore domain K+ channels as the only other choices, he said. The electrophysiology of the former does not match that of the current driven by the P12YR, hence Attwell concluded that he was chasing a two-pore domain channel. Analysis of published transcriptomes suggested microglia express only one gene of this kind, i.e., that encoding the two-pore domain halothane-inhibited K+ channel, aka THIK-1.
This channel sits in the cell membrane. It is widely expressed in mammalian tissue, including the brain where it is found mainly in microglial cells. It shares only 25-35 percent identify with other two-pore K+ channels. These channels have not been well-studied, but their regulation seems complex.
What might THIK-1 do in microglia? Ironically, Attwell found that it is not required for targeted motility; in other words, the cells still flood in to repair damaged tissue when that channel is blocked. But resting surveillance was another story. Blocking THIK-1 dramatically reduced extension and retraction of microglial projections in tissue slices, as well as the ground they managed to cover. The same occurred in vivo, said Attwell. Furthermore, the THIK-1 channel seems to be required for a key immune response of microglia, that is, their release of certain cytokines. Blocking THIK-1 prevented secretion of interleukin 1β in response to inflammatory signals.
Researchers at the meeting were both impressed by the data and concerned about its implications. This channel, and hence microglial baseline activity and immune response, can be blocked by gaseous anesthetics such as halothane and isoflurane, which are often used in research studies. “It’s hard to overstate how crucial that is,” noted Ransohoff. Researchers were concerned that microglial responses may be muted under anesthesia. “When we do experiments on live mice, particularly two-photon imaging, what can we use for anesthesia?” Ransohoff wondered.
Attwell reinforced another important theme that emerged at the meeting, namely that isolated microglia in vitro poorly mimic their brethren in the brain (see related Keystone news). Cultured microglia express little or no THIK-1, though perhaps they compensate with a different potassium channel or regulate motility in a different way, he suggested.—Tom Fagan
References
News Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.