Mutations
MAPT A152T
Quick Links
Overview
Pathogenicity: Alzheimer's Disease : Risk Modifier, Frontotemporal Dementia : Risk Modifier, Dementia with Lewy Bodies : Risk Modifier
Clinical
Phenotype: Alzheimer's Disease, Corticobasal Degeneration, Dementia with Lewy Bodies, Frontotemporal Dementia, Progressive Supranuclear Palsy, pallido-nigro-luysial atrophy variant of progressive supranuclear palsy, Other Tauopathy, Parkinson's Disease Dementia
Position: (GRCh38/hg38):Chr17:45991484 G>A
Position: (GRCh37/hg19):Chr17:44068850 G>A
dbSNP ID: rs143624519
Coding/Non-Coding: Coding
DNA
Change: Substitution
Expected RNA
Consequence: Substitution
Expected Protein
Consequence: Missense
Codon
Change: GCC to ACC
Reference
Isoform: Tau Isoform Tau-F (441 aa)
Genomic
Region: Exon 7
Research
Models: 3
Findings
Unlike the majority of pathogenic mutations in MAPT, the A152T variant does not appear to cause autosomal-dominant disease. Instead, it acts as a risk modifier and increases susceptibility to several neurodegenerative conditions, including Alzheimer's disease, frontotemporal dementia, and dementia with Lewy bodies. In addition to these conditions, A152T has been been found in people with progressive supranuclear palsy, corticobasal degeneration, and Parkinson's disease, although it has not been shown to affect risk in these conditions.
This variant was first reported in a 63-year-old man with an unclassified tauopathy characterized by progressive dementia with prominent speech impairment (Kovacs et al., 2011). His condition deteriorated over five years, and he developed myoclonus at end stage. His father had reportedly had similar symptoms, although segregation with disease could not be determined.
Another study identified this variant in four Caucasian individuals of northern European descent affected by difficult-to-classify tauopathy syndromes (Kara et al., 2012). The clinical picture in each case did not match typical disease presentations, making diagnosis difficult. One case was diagnosed with postencephalitic parkinsonism (PEP), although this was later revised to atypical tauopathy given the lack of encephalitis in her medical history. At age 26 she developed parkinsonism symptoms that lasted 54 years. She died at age 80. A family history of dementia was noted, but further details were not available. Neuropathological analysis confirmed an atypical tauopathy with neurofibrillary tangles, a mix of three-repeat and four-repeat tau isoforms, and a lack of TDP-43 pathology. A second unrelated mutation carrier developed a symmetrical akinetic rigid syndrome in her late 50s and was diagnosed with PD. Her disease lasted 10 years and did not involve cognitive impairment. At autopsy, her diagnosis was revised to pallido-nigro-luysial atrophy variant of progressive supranuclear palsy (PSP-PNLA). In this family, the mutation appeared to segregate with disease, being present in the proband's two symptomatic sisters and absent in her unaffected sister. A third proband case was diagnosed with idiopathic PD with dementia. He experienced symptom onset in his late 40s, starting with anxiety and concentration difficulties. Later he developed a stutter, depression, memory loss, bradykinesia, myoclonus, and visual hallucinations. He died seven years after onset and neuropathological analysis revealed a mix of neurofibrillary tangles and Lewy bodies consistent with a diagnosis of PD plus dementia. The final mutation carrier was diagnosed with corticobasal degeneration in the sixth decade of life. Disease in this case started with word-finding difficulties that progressed to aphasia and ultimately to muteness. This individual was also affected by unilateral rigidity and gait disturbances over an 11-year-disease duration. There was no documented family history of disease in this case.
The A152T variant was also described in two individuals of Spanish descent affected by AD (Jin et al., 2012). One case was confirmed by autopsy as having AD along with concurrent Lewy body pathology. Family history in this case was unknown. The other case is described as affected by sporadic early onset AD. The A152T variant appeared to segregate with disease in this case, as it was absent in an unaffected sibling.
In a large cohort, encompassing several independent series, the A152T variant was associated with an increased risk of FTD syndromes and AD (Coppola et al., 2012). Altogether, 15,369 subjects were screened, including those with FTD (n=2139), AD (n=3345), and controls (n= 9047). Although the precise degree of risk conferred by A152T is difficult to determine, it may increase risk of FTD nearly as much as APOE4 increases the risk of AD. The increased risk of AD associated with A152T is less than that associated with APOE4, but greater than for the other common risk variants such SORL1 and CLU. A follow-up study described the clinical histories and pathologies of nine A152T carriers, including those diagnosed with PSP (two), behavioral variant FTD (one), AD (two), non-fluent variant primary progressive aphasia (two), and corticobasal syndrome (two) (Lee et al., 2013).
The A152T variant has been described in a second case of PNLA (Graff-Radford et al., 2013). This individual had many symptoms consistent with PSP, including parkinsonism, postural instability, and downgaze supranuclear palsy, but also early and prominent eyelid apraxia, gait freezing, and micrography, all indicative of PNLA. This individual died at age 68 following a seven-year disease duration. Autopsy results supported the diagnosis of PNLA and strengthened the association of A152T with this rare tauopathy.
This variant also turned up in an ADNI study that sequenced AD patients with extreme biomarker levels in cerebral spinal fluid (Benitez et al., 2013). The study reported five heterozygous carriers of the A152T mutation in the cohort, one of whom had extremely low levels of Aβ42. This study noted that although the mutation occurred more frequently in the clinical cases than in controls, the association did not reach statistical significance in this cohort.
Recently, this variant was detected in one out of 188 individuals with Parkinson’s disease, one out of 188 individuals with PD with dementia, and two out of 376 controls (Schulte et al., 2015). The individual with PD had a negative family history and experienced symptom onset (resting tremor) at age 63. Later symptoms included bradykinesia, rigor, and postural instability. The individual with PD plus dementia experienced onset at age 64, also with resting tremor. Later symptoms included bradykinesia, rigor, postural instability, and dementia. Family history was unknown in this case. The two controls were Caucasian individuals from the KORA-Age cohort, based in Germany. In this study the A152T variant was reported as A469T, in reference to its position in the tau isoform with 776 amino acids (P10636-9).
Another recent paper detected this variant, reported as A469T, in an individual with apparently sporadic AD (Sala Frigerio et al., 2015). Clinical details were not reported.
In a large series of American and European people, the A152T variant was found to be associated with an increased risk of DLB, but not PD (Labbé et al., 2015). The variant was found in 10 out of 2456 controls (minor allele frequency 0.20 percent). Among PD patients, 18 out of 3229 carried the variant (MAF 0.28 percent) and amoung DLB patients, six out of 442 patients carried the variant (MAF 0.68 percent). In addition, two out of 181 patients with multiple system atrophy carried the variant (MAF 0.55 percent), a non-significant increase in frequency.
Neuropathology
Consistent with the variable clinical presentations associated with this variant, neuropathological reports are similarly diverse. Abnormal tau accumulation appears to be the unifying feature in all cases for which postmortem findings are available. In some cases prominent Lewy body pathology is seen (e.g., Kara et al., 2012). In other cases, the pathology is indicative of PNLA, as indicated by the prominent neuronal loss and tau deposition in the globus pallidus, subthalamic nucleus, and substantia nigra with lower levels of pathology in the motor cortex, striatum, pontine nuclei and cerebellum (Kara et al., 2012; Graff-Radford et al., 2013).
Biological Effect
This variant has been shown to impair tau's ability to bind microtubules, resulting in less-efficient microtubule assembly and impaired microtubule stability. In addition, although the mutant protein appears to aggregate with lower efficiency than wild-type protein overall, it is more prone to oligomer formation (Coppola et al., 2012). Isogenic human iPSCs generated from fibroblasts of an A152T carrier showed that the mutant tau is predisposed to proteolysis by caspases and other proteases and leads to greater tau pathology (Fong et al., 2013).
In silico, the A152T variant was predicted to be benign by PolyPhen2 (Sala Frigerio et al., 2015).
Last Updated: 18 Sep 2015
References
Paper Citations
- Kovacs GG, Wöhrer A, Ströbel T, Botond G, Attems J, Budka H. Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol. 2011 Jan-Feb;30(1):3-10. PubMed.
- Kara E, Ling H, Pittman AM, Shaw K, de Silva R, Simone R, Holton JL, Warren JD, Rohrer JD, Xiromerisiou G, Lees A, Hardy J, Houlden H, Revesz T. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol Aging. 2012 Sep;33(9):2231.e7-2231.e14. PubMed.
- Jin SC, Pastor P, Cooper B, Cervantes S, Benitez BA, Razquin C, Goate A, Ibero-American Alzheimer Disease Genetics Group Researchers, Cruchaga C. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer's disease Ibero-American cohort. Alzheimers Res Ther. 2012 Aug 20;4(4):34. PubMed.
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, Lane JR, Karydas AM, Kenet RO, Biernat J, Wang LS, Cotman CW, Decarli CS, Levey AI, Ringman JM, Mendez MF, Chui HC, Le Ber I, Brice A, Lupton MK, Preza E, Lovestone S, Powell J, Graff-Radford N, Petersen RC, Boeve BF, Lippa CF, Bigio EH, Mackenzie I, Finger E, Kertesz A, Caselli RJ, Gearing M, Juncos JL, Ghetti B, Spina S, Bordelon YM, Tourtellotte WW, Frosch MP, Vonsattel JP, Zarow C, Beach TG, Albin RL, Lieberman AP, Lee VM, Trojanowski JQ, Van Deerlin VM, Bird TD, Galasko DR, Masliah E, White CL, Troncoso JC, Hannequin D, Boxer AL, Geschwind MD, Kumar S, Mandelkow EM, Wszolek ZK, Uitti RJ, Dickson DW, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, , Ross OA, Rademakers R, Schellenberg GD, Miller BL, Mandelkow E, Geschwind DH. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet. 2012 Aug 1;21(15):3500-12. PubMed.
- Lee SE, Tartaglia MC, Yener G, Genç S, Seeley WW, Sanchez-Juan P, Moreno F, Mendez MF, Klein E, Rademakers R, Munain AL, Combarros O, Kramer JH, Kenet RO, Boxer AL, Geschwind MD, Gorno-Tempini ML, Karydas AM, Rabinovici GD, Coppola G, Geschwind DH, Miller BL. Neurodegenerative Disease Phenotypes in Carriers of MAPT p.A152T, A Risk Factor for Frontotemporal Dementia Spectrum Disorders and Alzheimer Disease. Alzheimer Dis Assoc Disord. 2013 Mar 25; PubMed.
- Graff-Radford J, Whitwell JL, Dickson DW, Josephs KA. Pallidonigroluysian atrophy associated with p.A152T variant in MAPT. Parkinsonism Relat Disord. 2013 Sep;19(9):838-41. Epub 2013 May 18 PubMed.
- Benitez BA, Karch CM, Cai Y, Jin SC, Cooper B, Carrell D, Bertelsen S, Chibnik L, Schneider JA, Bennett DA, , Fagan AM, Holtzman D, Morris JC, Goate AM, Cruchaga C. The PSEN1, p.E318G Variant Increases the Risk of Alzheimer's Disease in APOE-ε4 Carriers. PLoS Genet. 2013 Aug;9(8):e1003685. PubMed.
- Schulte EC, Fukumori A, Mollenhauer B, Hor H, Arzberger T, Perneczky R, Kurz A, Diehl-Schmid J, Hüll M, Lichtner P, Eckstein G, Zimprich A, Haubenberger D, Pirker W, Brücke T, Bereznai B, Molnar MJ, Lorenzo-Betancor O, Pastor P, Peters A, Gieger C, Estivill X, Meitinger T, Kretzschmar HA, Trenkwalder C, Haass C, Winkelmann J. Rare variants in β-Amyloid precursor protein (APP) and Parkinson's disease. Eur J Hum Genet. 2015 Jan 21; PubMed.
- Labbé C, Ogaki K, Lorenzo-Betancor O, Soto-Ortolaza AI, Walton RL, Rayaprolu S, Fujioka S, Murray ME, Heckman MG, Puschmann A, McCarthy A, Lynch T, Siuda J, Opala G, Rudzinska M, Krygowska-Wajs A, Barcikowska M, Czyzewski K, Sanotsky Y, Rektorová I, McLean PJ, Rademakers R, Ertekin-Taner N, Hassan A, Ahlskog JE, Boeve BF, Petersen RC, Maraganore DM, Adler CH, Ferman TJ, Parisi JE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Dickson DW, Ross OA. Role for the microtubule-associated protein tau variant p.A152T in risk of α-synucleinopathies. Neurology. 2015 Nov 10;85(19):1680-6. Epub 2015 Sep 2 PubMed.
- Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, Leung L, Ring KL, Seeley WW, Karydas A, Kshirsagar MA, Boxer AL, Kosik KS, Miller BL, Huang Y. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1(3):226-34. Epub 2013 Aug 29 PubMed.
- Sala Frigerio C, Lau P, Troakes C, Deramecourt V, Gele P, Van Loo P, Voet T, De Strooper B. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer's disease patients. Alzheimers Dement. 2015 Apr 29; PubMed.
Other Citations
Further Reading
Protein Diagram
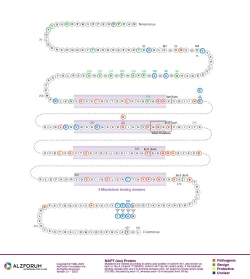
Primary Papers
- Kovacs GG, Wöhrer A, Ströbel T, Botond G, Attems J, Budka H. Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol. 2011 Jan-Feb;30(1):3-10. PubMed.
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, Lane JR, Karydas AM, Kenet RO, Biernat J, Wang LS, Cotman CW, Decarli CS, Levey AI, Ringman JM, Mendez MF, Chui HC, Le Ber I, Brice A, Lupton MK, Preza E, Lovestone S, Powell J, Graff-Radford N, Petersen RC, Boeve BF, Lippa CF, Bigio EH, Mackenzie I, Finger E, Kertesz A, Caselli RJ, Gearing M, Juncos JL, Ghetti B, Spina S, Bordelon YM, Tourtellotte WW, Frosch MP, Vonsattel JP, Zarow C, Beach TG, Albin RL, Lieberman AP, Lee VM, Trojanowski JQ, Van Deerlin VM, Bird TD, Galasko DR, Masliah E, White CL, Troncoso JC, Hannequin D, Boxer AL, Geschwind MD, Kumar S, Mandelkow EM, Wszolek ZK, Uitti RJ, Dickson DW, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, , Ross OA, Rademakers R, Schellenberg GD, Miller BL, Mandelkow E, Geschwind DH. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet. 2012 Aug 1;21(15):3500-12. PubMed.
Alzpedia
Disclaimer: Alzforum does not provide medical advice. The Content is for informational, educational, research and reference purposes only and is not intended to substitute for professional medical advice, diagnosis or treatment. Always seek advice from a qualified physician or health care professional about any medical concern, and do not disregard professional medical advice because of anything you may read on Alzforum.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.