Past Webinar
Together at Last, Top Five Biomarkers Model Stages of AD
Quick Links
Introduction
In this Webinar, Clifford Jack gave a slide talk summarizing a staged biomarker model of the Alzheimer’s cascade, followed by a panel discussion with Chet Mathis, David Holtzman, Henrik Zetterberg, Paul Aisen, Keith Johnson, Giovanni Frisoni, Douglas Galasko, and Larry Altstiel.
Please see comment by John Trojanowski.
- View/Listen to the Webinar
Click on this image to launch the recording.

Background
Background Text
By Gabrielle Strobel
The driving ambition of the past decade of Alzheimer disease research—human research, at least—has been the quest for validated biomarkers. But for most people, it was easy to lose track of a rapidly growing, sometimes contradictory literature. Which marker changes first? Second? Last? Which predicts disease? Tracks with cognitive decline? Flags if a drug works? Drug trials can’t employ all markers—how to choose? The answers boil down to how each marker matches up with stages of the disease. Clifford Jack, a leading MRI researcher at the Mayo Clinic in Rochester, Minnesota, has devoted much thought on integrating what is currently known about the five best-validated markers into a dynamic model of how things change over time on the long road to full-blown AD dementia. (Teaser tidbit: Aβ is first but far from be-all, end-all.) Jack presented his thinking at last summer’s ICAD conference in Vienna, Austria. There, it was uniformly welcomed as a fair-minded synthesis that captures an emerging consensus on how current biomarkers fit into the AD cascade. In the past, some scientists have naturally tended to favor the biomarker they’d been studying the most at their respective institutions. Representatives from each of these fields noted in Vienna that Jack’s model orders the markers objectively, moving the debate forward for everyone.
The model appeared as a “Personal View” essay in this month’s Lancet Neurology (Jack et al., 2010). Jack’s coauthors include clinicians David Knopman and Ron Petersen at Mayo; FDG-PET expert William Jagust at University of California, Berkeley; ADNI CSF core leaders Leslie Shaw and John Trojanowski of University of Pennsylvania in Philadelphia; ADCS leader Paul Aisen of UC San Diego; and ADNI chief Michael Weiner of UC San Francisco. Jack himself heads the MRI core of ADNI. In essence, their article states that of the five most studied fluid and imaging markers to date, Aβ changes first, as early as 20 years before symptoms appear. It also suggests that by the time a person has dementia, the disease has become quite disconnected from Aβ itself. MRI begins to change much later in the disease process, but then stays closely tied to it as dementia worsens. The authors place the synaptic dysfunction marker FDG-PET and the neurodegeneration marker CSF tau in between Aβ and MRI. The Alzforum editors recommend this article. For Webinar attendees without access to this journal, those without deep background in AD biomarker studies, or for those who simply had no time to absorb the 10-page article and its 115 citations, below is your editor’s summary.
The five biomarkers Jack et al. discuss include two for brain Aβ plaque deposition (CSF Aβ42 and PET amyloid imaging) and three for neurodegeneration (CSF tau, FDG-PET, and structural MRI). These markers are validated enough to be used in active therapeutic trials or large multisite observational studies such as ADNI. They have helped the field move past the black-and-white view of Alzheimer disease held some decades ago, when researchers thought, essentially, that people with AD pathology had dementia and people without AD pathology did not. This simple division started blurring when pathology studies revealed plaques and tangles in a sizable fraction of elders who had died with their cognition intact, and biomarker and longitudinal aging studies of the past 20 years subsequently swept aside this old binary view in favor of a more nuanced, dynamic picture. Now, the majority view holds that both pathologic and clinical changes occur gradually over time. Only after decades of early molecular change leading to neural dysfunction and degeneration, which in turn lead to clinical change and eventually functional loss, does the patient reach dementia as the end stage of the disease process. Importantly, the authors point out, each of these individual aspects develops on its own time course. At any given point in time, a given brain area may be at a different point along this process while the disease slowly eats its way through the brain.
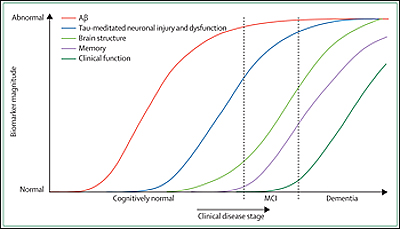
Image credit: Cliff Jack
Such a long process cries out for a sequential ordering, in other words, a way of staging Alzheimer disease by means of individual biomarker change. Clearly, individual biomarkers do not become abnormal and peak simultaneously, Jack et al. write.
Ordering biomarkers in time is important not only because it expresses the disease process in terms of a series of testable biological indicators, but also because clinical trial sponsors are searching for the best way to apply biomarkers for selecting patients and to measure drug effects. This is true both for a growing number of trials of disease-modifying drugs and for the concept of future prevention trials that is building steam across the field. Furthermore, understanding the temporal order of each biomarker then makes it possible to use a given marker for staging AD in vivo.
For background, Jack and colleagues first summarize what’s known about each of the five markers. They write that evidence from many labs strongly supports the notion that amyloid PIB-PET and low CSF Aβ42 are valid biomarkers for a person’s brain Aβ plaque load. Increased CSF tau is an indicator of tau pathological changes and neuronal injury, and it correlates with clinical disease severity. CSF tau is not specific for AD in that it also changes in stroke and brain trauma (e.g., after a boxing match) and, curiously, it does not change in pure tauopathies such as supranuclear palsy. FDG-PET measures brain metabolism and probably indicates synaptic activity. FDG-PET decreases in the AD pathogenic process from normal to AD in a regional pattern that matches affected areas and also correlates with cognitive impairment. According to Jack and colleagues, FDG-PET is a valid indicator of the synaptic dysfunction that accompanies neurodegeneration in AD. Last but not least, structural MRI measures cerebral atrophy as synapses disappear and neurons die. Like tau, it is not specific to AD, but it correlates closely with the severity of a person’s clinical impairment even late into the disease and with Braak staging and tangle pathology at autopsy. (It actually correlates for longer into the disease than CSF tau does, see below.)
So how to order these top five? In a nutshell, Jack and colleagues suggest that biomarkers of Aβ deposition become abnormal first, long before neurodegeneration and clinical symptoms appear. Biomarkers of neuronal dysfunction, injury, and neurodegeneration turn abnormal later. Cognitive symptoms are tied to biomarkers of this second category, not to biomarkers of Aβ deposition.
What are the main points of evidence the scientists cite for their model? On Aβ deposition happening first among the five markers, the authors note that autopsy, PIB-PET, and CSF Aβ42 studies all agree that up to 40 percent of cognitively normal elderly have a sufficient plaque burden to meet diagnostic criteria for AD. So amyloid alone does not equal dementia; however, it precedes and predicts dementia. By the time the first subtle symptoms appear some years later, all the other biomarkers have already turned as well. “There is strong evidence that MRI, FDG-PET, and CSF tau biomarkers are already abnormal in patients who are in the MCI phase of AD,” Jack and colleagues write. (This sentence appears to advance a related debate about the continued need for keeping mild cognitive impairment, or MCI, as a separate diagnostic category. In recent years, a shift away from using MCI for diagnosis in research settings has occurred along with the movement toward biomarkers [Dubois et al., 2007], particularly in Europe. While the present article also contains a statement about the continued usefulness of the MCI category and the term “dementia” in clinical practice, the article’s main thrust of staging the AD process by means of biomarker changes portrays the MCI stage of AD as part of a seamless disease continuum.)
The essay cites numerous studies, all supporting the conclusion that the abnormality of Aβ plaque biomarkers approaches a plateau before MRI atrophy and cognitive symptoms appear. After that point, the Aβ plaque load remains quite static. By and large, Aβ deposition is said and done before the person notices anything is amiss. In contrast, abnormalities in neurodegenerative biomarkers pick up speed as symptoms appear and from then on keep pace with a person’s cognitive decline. This is most strongly true for MRI.
In terms of what changes first and last, Aβ and MRI form the extremes, but how about the markers in between, i.e., CSF tau and FDG-PET? It is important to understand where on the disease continuum each of these is most dynamic, i.e., changes the most, and where it reaches its maximum, the authors note. On CSF tau, they cite a number of studies suggesting that this marker is most informative just prior to and during the earliest clinical symptoms, but—unlike MRI—no longer changes appreciably as the patient advances deep into dementia.
With regard to FDG-PET, the authors emphasize an interesting point. In general, each biomarker becomes abnormal on its own, non-linear time scale, and this puts in-vivo staging using biomarkers within reach. Beyond that general insight, FDG-PET in particular offers an additional advantage for staging in that it gives good anatomical resolution early on. Imaging abnormalities spread from one brain area to the next, such that at any given point in time, different brain areas are at different stages of disease. In other words, in any given area of a person’s brain there might be an amyloid phase, a neuronal dysfunction phase, an atrophy phase, and this will occur in an anatomical order. In this way, imaging is more informative than CSF, the authors write. Both FDG-PET and MRI volumetry give this staged anatomical information, but the former does so earlier in the disease process. The sigmoid curves of declining FDG-PET uptake take off first in the posterior cingulate, then in the lateral temporal, and later still in frontal areas. In graphs plotting biomarker abnormality across disease stage, these curves all originate to the left of—i.e., earlier than—otherwise similar regional MRI curves.
What does all this mean for clinical trials? Jack et al. confirm that selecting patients for trials of anti-amyloid drugs on the basis of Aβ biomarker evidence makes sense. One trial of prodromal AD is currently doing so; it requires abnormal CSF Aβ42 values and mild cognitive symptoms as inclusion criteria and excludes people with diagnosed dementia. More trials are being planned in similar ways. Biomarkers for neural dysfunction or neurodegeneration are not specific to AD and should not take precedence over Aβ markers as inclusion criteria, Jack et al. write. On the other hand, change in Aβ load relates poorly to improved cognition. Hence, it may hold little promise as an outcome measure of clinical importance beyond showing narrowly that the drug has engaged its target in the expected way. The “later” biomarkers could be used to show if the drug treats the neurodegeneration that marks the AD process, possibly as co-primary outcome measures along with the clinical ones that are in use today.
Any serious attempt at synthesizing a large literature into a central model is wont to draw criticism as well as praise, and the authors of this model hasten to point out some caveats themselves. For one, they write, they are fully aware that every published observation does not fit their model. For another, the model is generic, meaning there is room for different disease courses in individual people.
Moreover, ongoing research continues to generate data for a finer-grained separation of when which biomarker changes. For example, early indications are that CSF Aβ precedes PIB-PET by a bit; likewise, data emerging from ADNI suggest that the FDG-PET curve might begin to take off earlier, i.e., lie to the left, of the CSF tau curve.
Importantly, the model has big gaps where robust biomarkers simply do not exist yet. Would if there were chemical biomarkers for Aβ oligomers or imaging markers for diffuse amyloid deposits! Similarly, the field urgently awaits PET ligands for tau abnormalities, microglial activation, and α-synuclein deposits, the latter to assess overlapping pathologies on the spectrum from AD to Parkinson disease. Seeing TDP-43 deposits with PET scanning would be nice, too. “Thus, our biomarker model…is just that—a model of the stages of disease that can be assessed with currently validated biomarkers, and not a comprehensive model of all pathological processes in AD,” the authors acknowledge.
They do, however, point a finger at one widely accepted notion that is simply not borne out by this biomarker model of AD. A much-cited postmortem study that describes entorhinal neurofibrillary tangles as preceding cortical plaques (Braak and Braak, 1997; Braak and Braak, 1991) has led to the conclusion that tangle accumulation is the initiating event in AD (Duyckaerts and Hauw, 1997). This view finds no support in AD genetics, either, the authors write. On the question of whether Aβ or tau becomes abnormal first in humans, the answer appears to be in.
Other observations do not fit the model at present but may need to be incorporated as scientists learn more. For example, some PET studies show abnormally low FDG uptake in cognitively normal ApoE4 carriers even at young adult ages, decades before AD symptoms would be expected (Reiman et al., 2004; Scarmeas et al., 2003). It’s a puzzle at present if this is a developmental consequence of growing up with ApoE4 or a very early harbinger of AD. Likewise, other imaging modalities may find a place in this model as they mature and are becoming more widely used, for example, connectivity imaging using functional MRI, the authors write.
In all, the new model offers scientists a rich framework for testing various hypotheses. For example, because ApoE4 carriers tend to get AD earlier, the sigmoid curves of their biomarker changes should be shifted to the left (i.e., earlier in time) than those of non-carriers. Also, modifying factors that lengthen the lag time between Aβ accumulation and symptoms, such as cognitive reserve, should spread the Aβ and neurodegeneration curves farther apart on the time axis; this would represent people who function well for quite some time despite having a head full of amyloid. Au contraire, factors thought to shorten this lag time, such as cerebrovascular disease, would be expected to move the curves closer together; this would represent people in double trouble who become symptomatic soon after amyloid settles in their brains. Through studies that combine genotyping with longitudinal biomarker panel measurements, for example, ADNI, genes influencing AD risk can be tested for whether they affect this lag phase as well.
References
Paper Citations
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010 Jan;9(1):119-28. PubMed.
- Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, Meguro K, O'brien J, Pasquier F, Robert P, Rossor M, Salloway S, Stern Y, Visser PJ, Scheltens P. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007 Aug;6(8):734-46. PubMed.
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997 Jul-Aug;18(4):351-7. PubMed.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
- Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak's stages in the general population: can we know?. Neurobiol Aging. 1997 Jul-Aug;18(4):362-9; discussion 389-92. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):284-9. PubMed.
- Scarmeas N, Habeck CG, Stern Y, Anderson KE. APOE genotype and cerebral blood flow in healthy young individuals. JAMA. 2003 Sep 24;290(12):1581-2. PubMed.
Other Citations
{kind=link}
External Citations
Further Reading
Papers
- Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000 Apr 14;288(5464):335-9. PubMed.
Panelists
-
 Clifford R. Jack, M.D.
Mayo Clinic
Clifford R. Jack, M.D.
Mayo Clinic
-
 Chester Mathis, PhD
PET Facility
Chester Mathis, PhD
PET Facility
-
 David Holtzman, B.S., M.D.
Washington University
David Holtzman, B.S., M.D.
Washington University
Current Scientific Advisory Board Member -
 Henrik Zetterberg, M.D., Ph.D.
University of Gothenburg
Henrik Zetterberg, M.D., Ph.D.
University of Gothenburg
-
 Paul Aisen, MD
USC Alzheimer’s Therapeutic Research Institute
Paul Aisen, MD
USC Alzheimer’s Therapeutic Research Institute
-
 Keith Johnson, M.D.
MGH, BWH, HMS
Keith Johnson, M.D.
MGH, BWH, HMS
-
 Giovanni Frisoni, MD
IRCCS Fatebenefratelli
Giovanni Frisoni, MD
IRCCS Fatebenefratelli
-
 Douglas Galasko, M.D.
University of California, San Diego
Douglas Galasko, M.D.
University of California, San Diego
Comments
Yale University School of Medicine
Some thoughts on this interesting proposal:
This article raises important questions regarding our ability to monitor the pathogenesis of AD. How can we detect the earliest changes in the human brain that will eventually lead to clinical dementia? Where do these changes start, what processes are involved, is progression linear or episodic, or even reversible? None of these questions can be answered at the present time, but the findings described here represent a promising beginning.
They confirm what others have shown: that Aβ deposits are, unfortunately, an all too common feature of late adult life, but they, acting alone, may not be enough to provoke the neurodegenerative changes that result in dementia. Other factors, including neurofibrillary tangles, must play a role, but how they do is still a mystery. The most informative, and, I think, horrifying idea proposed by the lead author in his recent study (Jack et al., 2009) is the hypothesis that “amyloid deposition in patients with typical late-onset Alzheimer disease would have to begin in subjects in their forties.”
My question is this: Are the five markers that have been described here capable of detecting the early stages of the disease? A related question is whether they can be used to measure rates of progression of disease in younger patients at a stage in the disease where therapeutic intervention is a realistic possibility. The markers discussed in this report are entirely based on the presumed consequences of Aβ activity. In this regard other markers that reflect processes that might contribute to Aβ toxicity may be more informative. No mention is made of markers that might reflect concurrent inflammation and oxidative damage, both of which are likely to enhance the amyloid effects, as Golde and others have repeatedly stressed.
References:
Jack CR, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC, . Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009 May;132(Pt 5):1355-65. PubMed.
Washington University
Dr. Jack and others offer a well-reasoned and clearly explained model in which five of the “most well-validated” Alzheimer disease (AD) biomarkers provide complementary information that might be used as a panel to identify three stages of AD progression: presymptomatic, mild cognitive impairment (MCI), and dementia.
This model reflects what appears to be a growing consensus in the AD biomarker field, as expressed in other recent reviews, including our own (Perrin et al., 2009), that amyloid-β deposition in the brain (identifiable by low CSF levels of Aβ42 and generally by PET-PIB signal above a certain threshold) occurs very early in the presymptomatic phase, and is followed many years later by neurodegeneration (tau pathology and synaptic and neuronal loss), measurable by CSF tau and phospho-tau, structural MRI, and FDG-PET.
We hope this review, focused as it is on current “shovel-ready” biomarkers, might stimulate a more widespread application of biomarker science to AD clinical trial design and monitoring. Such a practice would undoubtedly increase statistical power, shorten duration, and decrease the necessary size and cost of such trials. Perhaps most importantly, the use of such biomarker data might allow clinical trials to focus attention on presymptomatic/preclinical stages of the disease, when disease-modifying therapies seem likely to have a much better chance to prevent the synaptic and neuronal loss associated with cognitive dysfunction in AD. As noted in this work, some argue that past trials of “plaque-clearing” treatments failed because they were administered after significant brain damage had already occurred.
We would like to emphasize, however, (and applaud the authors for mentioning) that the great promise shown by the five biomarkers discussed in this review should not inspire complacency. The sensitivity and specificity of these markers for diagnosis and prognosis, though quite impressive, are not perfect. In the near future, this model might be improved by the addition of other biomarkers that are currently in earlier stages of validation. As acknowledged by the authors, entire facets of well-recognized AD pathology—in particular, direct measures of amyloid-β oligomers, neurofibrillary pathology, oxidative damage, and inflammation (microglial activation and reactive astrocytosis)—are not yet represented in the model.
Nevertheless, the allure of future innovations should not discourage the application of excellent technology today. We echo the notions described in this article, and strongly encourage the incorporation of these concepts in new clinical trial design.
References:
Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009 Oct 15;461(7266):916-22. PubMed.
Arizona Alzheimer's Consortium
In this important article, Cliff Jack and his colleagues have done a wonderful job of synthesizing brain-imaging and cerebrospinal fluid (CSF) data to characterize and compare the trajectory of biomarker changes in the presymptomatic and progressively symptomatic stages of Alzheimer disease (AD).
Based on available findings, they suggest that 1) the sequence of biomarker changes begins with progressive evidence of amyloid-β (Aβ) pathology and continues with evidence of phospho-tau and total tau pathology and regional hypometabolism in the presymptomatic stages of AD; 2) the sequence continues with evidence of accelerated brain tissue atrophy, which heralds the onset of cognitive decline; and 3) measures of amyloid pathology appear to plateau, while brain-imaging measures of metabolic decline and brain atrophy continues to progress during the symptomatic stages of AD. They suggest that the trajectory of biomarker changes is likely to be non-linear, and that the temporal sequence of brain changes in different brain regions is likely to follow a characteristic pattern, roughly corresponding to the regional progression of tau pathology and synaptic and neuronal loss. They describe several practical implications of this sequence for the emerging role of biomarkers in the diagnosis of AD and the evaluation of AD-modifying treatments. They also note some of the proposed model’s caveats and limitations.
With all due respect to Occam’s razor, I’d like to discuss a few caveats, some of which Dr. Jack and his colleagues acknowledged during the thoughtful presentation of their theoretical model:
1. While the authors’ model suggests that certain Aβ species play the earliest role in the predisposition to AD, triggering the cascade of neuropathological events, there is reason to believe that even earlier brain changes may be involved in the predisposition to detectable Aβ alterations and AD:
While I realize Occam might not approve of the idea, it is possible that certain measures of synaptic or tau pathology may be involved in the earliest predisposition to late-onset AD. They may conspire with other age-related changes in the development of toxic Aβ species, which in turn lead to the proposed cascade including progressive neuropathology and synaptic and neuronal loss. It is possible that early onset AD is different in that it begins with the overproduction of certain Aβ species.
2. The trajectory of brain changes may be even more complex than the authors propose, at least in some brain regions. For instance, there may be preferential declines in frontal cortex related to brain aging, beginning in a person’s forties, and a subsequent acceleration in these frontal cortex changes in the later stages of AD. Speaking of frontal cortex, it would be very helpful to know why the frontal cortex is relatively spared from brain atrophy, metabolic decline, and executive dysfunction despite the presence of significant fibrillar Aβ in the presymptomatic and early symptomatic stages of late-onset AD—and why the striatal neurons appear to be relatively spared despite unusually high Aβ accumulation in the presymptomatic and symptomatic stages of early onset AD.
3. I agree with the proposition that biomarkers may help to select symptomatic and presymptomatic clinical trial participants in the evaluation of putative AD-modifying treatments. But I would also suggest a potential caveat: While very high Aβ pathology or high CSF tau/Aβ42 measurements may predict subsequent rates of progression to symptomatic AD, they may also predict a presymptomatic stage in which the initiation of an Aβ-modifying treatment proves to be too little, too late for the treatment to exert a profound effect. An alternative at-risk enrichment strategy that we have been exploring is the selection of individuals at increased genetic risk for early or late-onset AD close to their anticipated median age at clinical onset in presymptomatic AD trials. While a limitation of this approach may be generalizability to non-carriers of these genes, one advantage would be the inclusion of at-risk individuals with higher or lower levels of Aβ or tau pathology, providing information about whether the group with greater or lesser pathology is preferentially responsive to the proposed treatment.
However it is accomplished, my colleagues and I are especially interested in the idea of evaluating investigational AD-modifying treatments with sufficient safety and tolerability data in cognitively normal people at highest imminent risk of symptomatic AD in order to 1) conduct presymptomatic AD trials earlier than otherwise possible because of a more favorable benefit-to-risk ratio; 2) determine the extent to which a clinically proven treatment’s different biomarker effects predict a clinical benefit, thus providing some of the evidence needed to approve future presymptomatic treatments based on reasonably likely surrogate biomarker endpoints; 3) provide a better test of the amyloid hypothesis than clinical trials of Aβ-modifying treatments in symptomatic patients; and 4) give these at-risk participants access to some of the most promising investigational treatments in presymptomatic clinical trials.
Lund University
Reading this story, like so many others on Alzforum, made me think of how intraneuronal Aβ accumulation can be pertinent also to this discussion. Long a fringe topic, this issue has been accumulating evidence over the past decade, and delay in considering it could hold back efforts to better diagnose and treat this complex disease. Although rarely mentioned, reduced CSF Aβ42 as the earliest harbinger of future AD fits well with the scenario of aberrant accumulation of β amyloid within neurons in the brain. Many groups by now have shown that early intraneuronal Aβ accumulation precedes plaques and tangles, begins in the most vulnerable AD brain areas, and has been directly associated with cognitive decline and synaptic damage (for review, see Gouras et al., 2005; Laferla et al., 2007).
A message that comes out of this discussion is that Aβ plays an early role in AD but is not the immediate cause of degeneration, since cognitive decline develops after the drop of CSF Aβ42 and even after plaques on brain imaging. The neurologists among us know that a person can have considerable brain pathology in the absence of cognitive deficits on bedside testing. Examples are a grapefruit-size tumor in the frontal lobes, where you could doubt your competence as a clinician when you can’t demonstrate any deficits on exam in front of anticipating residents and medical students. Similarly, relatively large strokes in certain regions of the brain do not always show deficits. Furthermore, most elderly individuals have evidence of some cerebrovascular disease/damage on brain MRI, although most of them don’t have neurological dysfunction. Thus, pathological intraneuronal Aβ accumulation and Aβ plaques still can be (and by electron microscopy, indeed, are) signs of brain damage even when cognitive testing does not pick up dysfunction.
I look forward to intraneuronal Aβ entering discussions such as this, although this by no means precludes other topics being important next to Aβ, from ApoE to tau, oxidative stress to inflammation, etc. Using the atherosclerosis analogy, cholesterol also does not define all of cardiovascular disease. Continuing the analogy, and in light of the fear expressed by Dr. Marchesi, atherosclerosis begins even earlier than one's forties, yet here progress in therapy has been astounding.
References:
Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005 Oct;26(9):1235-44. PubMed.
Laferla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007 Jul;8(7):499-509. PubMed.
Make a Comment
To make a comment you must login or register.