Studies Point to DNA Difficulties in ALS/FTD
Quick Links
Three studies published this week on the etiology of amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) spotlight a cell compartment that is often overlooked. They focus squarely on the nucleus, specifically the integrity of its DNA. In two papers, Leonard Petrucelli at the Mayo Clinic, Jacksonville, Florida, and colleagues dissect the leading genetic cause of ALS/FTD, a repeated hexanucleotide expansion in the C9ORF72 gene. The expansion gives rise to two abnormal RNAs encoding five different dipeptide repeat proteins that aggregate in cells. In the February 15 Science, Petrucelli and co-workers show the most toxic of those poly-dipeptides homes to the nucleus in mice and in people with ALS/FTD, where it disrupts the structure of chromatin and revs up transcription of normally silenced stretches of repetitive DNA. The resulting RNAs contribute to neurodegeneration. In their second paper, in the February 15 Molecular Neurodegeneration online, they proffer a new mouse model of ALS/FTD that expresses a construct carrying 149 hexanucleotides. The mice show many hallmarks of ALS/FTD, including accumulating stress granules, cytoplasmic TDP-43 deposits, poly(PR) inclusions, and nucleocytoplasmic transport deficits. The third study, published February 11 in PNAS, reports a role for TDP-43 in DNA repair. Muralidhar Hegde and colleagues at the Houston Methodist Research Institute, Texas, show that with loss of nuclear TDP-43 in diseased neurons, double-strand DNA breaks accumulate, which contribute to neurodegeneration.
- In ALS/FTD model of C9ORF72 toxicity, poly(PR) targets nucleus, alters chromatin structure and function.
- TDP-43 pathology hampers DNA repair.
- Nuclear pathways may offer novel therapeutic targets.
“The studies are intriguing, and bring together some different lines of research. They show a growing interest in events occurring in the nucleus, which neuroscientists have not focused on so much before,” said Ben Wolozin, Boston University.
“The Science paper’s provocative finding raises a new pathway that could contribute to C9-FTD/ALS. One note of caution is that the relevance to the disease in human patients is not currently clear, and no doubt will be a major focus of future work,” wrote Adrian Isaacs, University College London, to Alzforum.

Nuclear Meltdown? In mice expressing poly(PR), the peptides (green) collect in the nucleus, and induce deformation of the nuclear lamina (red), a linchpin of chromatin structure. [Zhang et al., Science 2019.]
Chromatin Crisis
Of all the dipeptide repeat proteins expressed by the C9ORF72 expansion, the proline-arginine, poly(PR), and glycine-arginine poly(GR) polymers cause the most havoc in cultured cells and in fruit fly models of neurodegeneration. However, poly(PR) is barely detectable in ALS/FTD, so researchers were unsure if it contributes to disease. To find out, first author Yong-Jie Zhang in Petrucelli’s lab injected newborn mice intraventricularly with adenoviruses to induce expression of a 50-repeat poly(PR) conjugated to green fluorescent protein. GFP-(PR)50 was indeed extremely toxic: Sixty percent of poly(PR)-injected mice died within four weeks, having failed to gain weight or develop their brains normally. Mice that survived longer lost neurons and brain weight over the next six months, developing neuroinflammation, motor deficits, and memory impairment. The number of poly(PR)-positive neurons in the cortex and cerebellum diminished with time, suggesting the peptide kills neurons, Petrucelli told Alzforum. “That may explain why PR deposits are rare in postmortem brain tissue from people with C9-FTD/ALS who had had end-stage disease,” he said.
Previous work from Petrucelli’s lab and others indicated that a different dipeptide, poly(GR), targeted ribosomes and inhibits protein synthesis (Jun 2018 news; Jun 2018 news). But poly(PR) was different: the protein exclusively turned up in the nucleus in neurons, disrupting the nuclear lamina and associating with histones and DNA—especially condensed, transcriptionally silent heterochromatin, which mostly comprises repetitive DNA sequences. Poly(PR) altered histone methylation and reduced levels of the heterochromatin protein 1a (HP1α), which participates in gene silencing, and transcription of repeat heterochromatin sequences promptly surged. Those transcripts formed double-stranded RNA, which, in mammalian cells, signal viral infection and trigger innate immune responses. “Obviously, that’s not a good thing for cells,” said Petrucelli. When Zhang knocked out HP1α in cultured neurons, double-stranded RNA increased, along with activated apoptotic caspase 3.
The researchers saw the same localization of poly(PR) with histones in brain tissue from people with ALS/FTD caused by C9ORF72 expansion. Others have already noted accumulation of double-stranded RNA in such samples (Prudencio et al., 2017).
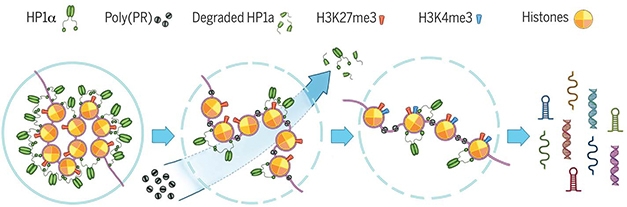
Bad PR. Poly(PR) binds to condensed heterochromatin, displacing the silencing factor Hp1α and altering histone methylation. This loosens chromatin packing and opens repetitive DNA elements to transcription, leading to accumulation of double-stranded RNAs in cells. [Zhang et al., Science 2019.]
Like many RNA binding proteins that have been linked to ALS/FTD pathology, HP1α undergoes liquid-liquid phase separation, forming protein droplets that sequester heterochromatin and promote gene silencing. Poly(PR) and other DPRs congeal liquid droplets of RNA-binding proteins (Oct 2016 news). When Zhang added poly(PR) to preformed HP1α liquid droplets in vitro, the droplets developed solid specks inside, and then burst [see video below]. If this happened in vivo, poly(PR) could evict Hp1α from the DNA, opening the DNA up to transcription, the authors speculate.
Burst Bubbles: Liquid-phase HP1α droplets curdle and rupture when exposed to poly(PR) in vitro (top movie) but not when poly(PA) is added (bottom movie). [Movies courtesy Zhang et al., Science 2019.]
Trouble with TDP-43
While poly(PR) relocated nucleocytoplasmic transport proteins, which have been linked to TDP-43 accumulation, it was insufficient to provoke cytosolic translocation and aggregation of TDP-43 itself, a major hallmark of ALS/FTD (Oct 2018 news). “Consistent with our previous work, formation of TDP-43 cytoplasmic inclusions seems to require interaction between multiple dipeptide repeat proteins,” said Petrucelli.
TDP-43 dislocalization and aggregation did occur in their C9ORF72 expansion model. In the Molecular Neurodegeneration paper, joint first authors Jeannie Chew and Casey Cook report that mice expressing the 149 repeat C9ORF72 construct recapitulate all the key pathologies of C9-ALS/FTD. They accumulate both sense and antisense RNA, translate the full complement of dipeptide repeat proteins, deposit extensive cytoplasmic phospho-TDP-43 inclusions, and suffer gliosis, neurodegeneration, and cognitive and motor defects. The lab’s previous model, a 66-repeat mouse (Chew et al., 2015) expressed only C9ORF72 sense RNA, which encodes poly(GP), (GA), and (GR). The antisense RNA produces poly(PA) and (PR) proteins, too. As they age, the new 149-repeat mice accumulate the DPRs in amounts that mirror ALS/FTD cases. “GP and GA are most abundant, followed by GR, then PA and PR, consistent with what we find in human disease,” Petrucelli said. The mice offer a complete model to test potential therapies aimed at different pathways, he said.
“The new 149 GGGGCC repeat AAV model improves on the original 66-repeat model, particularly in the expression of antisense repeats and the more robust development of cytoplasmic pTDP-43 pathology. This will be a powerful model for future investigation of disease mechanisms and methods to ameliorate the disease,” Isaacs concurred.
In the 149-repeat mice, stress granule proteins mislocalize early, by three months of age, before TDP-43 deposition begins. This supports the idea that stress granules drive TDP-43 proteinopathy, Petrucelli said.
In the PNAS paper, first author Joy Mitra outlined how TDP-43 mislocalization to the cytosol contributes to neuronal cell death by preventing timely repair of DNA damage. Best known for binding RNA, TDP-43 also associates with the DNA repair protein Ku, and there have been hints it participates in DNA repair (Freibaum et al., 2010; Hill et al., 2016; Yu et al., 2012).
Mitra found that in human motor neurons and neural progenitors in culture, TDP-43 associated with DNA repair enzymes. TDP-43 was rapidly recruited to damaged chromatin in neurons or neuroblastoma cells treated with the topoisomerase blocker etoposide, and it remained there until repair was complete. Etoposide promotes double-stranded breaks that topoisomerases make while untangling DNA. In SH-SY5Y neuroblastoma cells, CRISPR/Cas9-mediated depletion of TDP-43 led to accumulation of these breaks, which triggered apoptosis via caspase-3. In spinal tissue from people with sporadic ALS, extranuclear TDP-43 pathology correlated with DNA damage, defects in double-strand repair, activation of DNA damage response signaling pathways, and neurodegeneration. The authors suggest that therapies targeted to DNA repair therapies may be helpful to prevent cell death in ALS. Petrucelli told Alzforum his lab is examining the 149-repeat mouse model for signs of DNA damage.–Pat McCaffrey
References
News Citations
- In New ALS/FTD Mouse Model, Poly(GR) Peptides Poison Ribosomes
- Dipeptide Repeats May Hobble Ribosomes in C9ORF72-FTD Patient Brain
- ALS Research ‘Gels’ as Studies Tie Disparate Genetic Factors Together
- Dipeptide Repeat Proteins Trigger TDP-43 Pathology, Faulty Nuclear Import
Paper Citations
- Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, Ebbert MT, van Blitterswijk M, Zhang YJ, Jansen-West K, Baker MC, DeTure M, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, Link CD. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet. 2017 Sep 1;26(17):3421-3431. PubMed.
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015 Jun 5;348(6239):1151-4. Epub 2015 May 14 PubMed.
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010 Feb 5;9(2):1104-20. PubMed.
- Hill SJ, Mordes DA, Cameron LA, Neuberg DS, Landini S, Eggan K, Livingston DM. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci U S A. 2016 Nov 29;113(48):E7701-E7709. Epub 2016 Nov 14 PubMed.
- Yu Z, Fan D, Gui B, Shi L, Xuan C, Shan L, Wang Q, Shang Y, Wang Y. Neurodegeneration-associated TDP-43 Interacts with Fragile X Mental Retardation Protein (FMRP)/Staufen (STAU1) and Regulates SIRT1 Expression in Neuronal Cells. J Biol Chem. 2012 Jun 29;287(27):22560-72. PubMed.
Further Reading
Primary Papers
- Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, O'Raw AD, Pickles SR, Prudencio M, Carlomagno Y, Gachechiladze MA, Ludwig C, Tian R, Chew J, DeTure M, Lin WL, Tong J, Daughrity LM, Yue M, Song Y, Andersen JW, Castanedes-Casey M, Kurti A, Datta A, Antognetti G, McCampbell A, Rademakers R, Oskarsson B, Dickson DW, Kampmann M, Ward ME, Fryer JD, Link CD, Shorter J, Petrucelli L. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019 Feb 15;363(6428) PubMed.
- Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, Castanedes-Casey M, Kurti A, Stankowski JN, Disney MD, Rothstein JD, Dickson DW, Fryer JD, Zhang YJ, Petrucelli L. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019 Feb 15;14(1):9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.