At Risk, or Already Alzheimer’s? Elevated Aβ Predicts Cognitive Decline
Quick Links
By many estimates, around a third of people in their golden years appear cognitively healthy, but actually harbor a mess of amyloid plaques in their brain. Are they merely at risk of developing Alzheimer’s disease, or have they already entered its early stages? A new study provides additional support for the latter scenario. Drawing on longitudinal data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI), researchers led by Michael Donohue and Paul Aisen at the University of Southern California in San Diego measured brain amyloid in cognitively normal people at baseline, then tracked their cognitive performance and other measures for up to a decade. As reported in JAMA on June 13, people who had elevated amyloid from the get-go ultimately slipped on cognitive tests, while those who started with normal Aβ tended to maintain their performance over the following years. The brain shrank in both groups, but did so faster in those who had started the study with elevated Aβ.
The findings stop short of nailing down a causal link between amyloid deposition and subsequent cognitive decline. However, as noted in an accompanying editorial by Pieter Jelle Visser and Betty Tijms of VU University Medical Center in Amsterdam, the study “clearly indicates that amyloid pathology in cognitively normal older persons is not a benign phenomenon of normal aging but part of a progressive neurodegenerative disease.” This has several profound implications, they added, including the idea that if and when an amyloid-targeting drug becomes available, asymptomatic people with elevated amyloid could be among those who receive it.
Aβ can start accumulating in the brain two decades before cognitive symptoms of AD emerge. Postmortem studies have shown that a significant proportion of people over 65 had elevated amyloid at the time they died but had shown no outward cognitive symptoms (see Nov 2012 news; May 2015 news). Elevated Aβ drives up risk for cognitive decline, especially when other markers of neurodegeneration are present (Sep 2013 news; Jack et al., 2016; Insel et al., 2016). However, given the long preclinical phase of the disease, it has been challenging for researchers to cinch a causal link between Aβ deposition and subsequent clinical manifestations of AD.
“In science, we tend to reserve the ‘C word’ for situations where we can randomize people. Failing that, we look for temporal precedence,” Donohue told Alzforum. Given the obvious impossibility of randomizing people to accumulate amyloid, Donohue and colleagues did the next best thing: They tracked cognition and markers of neurodegeneration in a group of 445 cognitively normal people, just under half of whom had started the study with elevated Aβ.
Baseline assessments for some ADNI volunteers started in mid-2005. The researchers tracked participants for an average of 3.1 years, with a maximum follow-up of just over 10 years. The participants averaged 74 years of age. At baseline, Aβ status was assessed either via florbetapir-PET scan, PiB-PET scan, or CSF Aβ42. In all, 202 participants had elevated amyloid at baseline, as indicated by a standardized uptake value ratio above 1.1, or CSF Aβ42 concentration below 192pg/mL. Six months later and once a year thereafter, the participants underwent further Aβ measurements, MRI scans to assess brain volume and metabolism, and a battery of cognitive tests. These included the mini mental state exam (MMSE), the Clinical Dementia Rating Sum of Boxes (CDR-SB), the Logical Memory Delayed Recall test, and a modified version of the Preclinical Alzheimer Cognitive Composite (PACC).
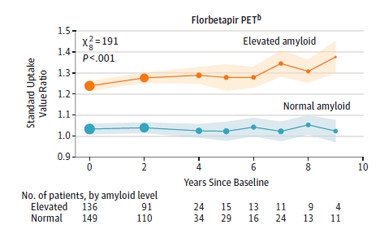
Bimodal Beginning.
Participants started ADNI with little middle ground in terms of Aβ accumulation as measured by amyloid-PET scans. [Courtesy of Donohue et al., JAMA 2017.]
Strikingly, even at baseline, participants were partitioned squarely on either side of the amyloid divide, with few people lingering near the threshold of elevated Aβ. Over the following decade, brain Aβ levels held steady in the normal group, and slightly increased in the elevated group. Few people crossed the threshold during follow-up, Donohue said. This bimodal distribution at baseline and beyond indicates that most people had jumped from normal to elevated Aβ status prior to the baseline measurements, and that “people are either on the AD trajectory, or they’re not,” he said. The finding also suggests that, given the approximately 20-year lag period between abnormal Aβ and cognitive symptoms, few people in the normal Aβ group are likely to develop symptomatic AD in their lifetimes, he said.
Levels of CSF tau and phospho-tau were also measured in a subset of participants, for up to six years. Like Aβ, tau partitioned between the normal and elevated Aβ groups at baseline, with concentrations of tau and p-tau far higher in the elevated Aβ group. Tau and p-tau continued to increase in both groups over the observation period, plateauing at around six years.
How did the two Aβ groups compare in terms of cognitive performance? Scores between the two were neck-and-neck for a while, but by four years, people in the elevated Aβ group performed significantly worse on the PACC, MMSE, and CDR-SB than those in the normal group, with ApoE4 carriers suffering the steepest decline. By this time, nearly a third of elevated-Aβ participants had developed symptoms consistent with prodromal AD, 14.5 percent with early MCI, and four percent with late MCI.
Roughly half as many people in the normal Aβ group had hit these progression events. Donohue attributed their progression either to their having had sub-threshold Aβ accumulation, or to suspected non–Alzheimer disease pathophysiology (SNAP). The findings mesh with another recent ADNI data analysis, in which Aβ deposition-negative people with MCI had lower CSF tau, less brain atrophy, a lower frequency of ApoE4, and slower clinical progression than did their counterparts who did have brain Aβ deposition (Schreiber et al., 2017).

In the current study, compared to around 180 people who had been followed for at least four years, only 18 volunteers had reached the 10-year mark when the data were analyzed, meaning that a small group drove those eight- and 10-year time points. The researchers used those measurements along with data from earlier years to estimate that 88 percent of people in the elevated Aβ group would progress beyond the prodromal stages of AD in the coming decade, compared to just 29 percent of those in the normal Aβ group, for a threefold increase in risk.
Evidence of neurodegeneration also tracked with baseline Aβ status. In addition to the elevated concentrations of CSF tau/p-tau in the elevated Aβ group, the researchers found that both total brain and hippocampal volume shrank most significantly in those volunteers.
Samantha Burnham of the Commonwealth Scientific and Industrial Research Organization in Canberra, Australia, found it interesting that while baseline levels of Aβ and other biomarkers differed between the normal and elevated Aβ groups, they had similar profiles throughout follow-up. “[This suggests] that decline on clinical and cognitive measures is associated with abnormal pathology, but not necessarily increasing pathology,” she wrote to Alzforum. She also wondered whether ApoE4 carriers declined most steeply simply because they actually had higher levels of baseline Aβ than other people within the elevated Aβ group.
ADNI researchers will continue to track participants, with the aim of further clarifying the relationships between Aβ deposition, tau pathology, neurodegeneration, and cognitive decline. Doing so will be essential to inform the optimal design of secondary prevention trials, commented Eric Reiman of Banner Alzheimer’s Institute in Phoenix, who commended the work.
“Although the study design does not allow for drawing conclusions on causality, the data show that amyloid positivity is very common and should not to be neglected,” commented Henrik Zetterberg at the University of Gothenburg in Sweden. “The study [also] shows we have tools to detect the group that might benefit from amyloid-targeting treatments, which will be clinically important once such drugs are available.”
Why does it matter whether elevated Aβ in cognitively normal people is a risk factor for AD or an early part of the disease process? The distinction indeed has implications that go beyond semantics, the authors pointed out. Obtaining an early diagnosis following a positive amyloid scan may help individuals and families prepare for the blow of the disease, although many researchers still advise against screening people for elevated Aβ in the absence of an approved Aβ-targeted drug. Once a drug is approved, treatment of cognitively healthy people with elevated amyloid would be considered disease management, rather than risk reduction. This distinction would likely have broad implications, ranging from amyloid screening, decisions to begin treatment, and the cost and coverage of drugs.
The researchers further estimated that accounting for people in the preclinical and prodromal stages of AD would more than double prevalence estimates of the disease worldwide.—Jessica Shugart
References
News Citations
- API Echoes DIAN: Biomarker Changes Precede Symptoms by 20 Years
- Meta-Analyses Deliver Most Definitive Data Yet on Amyloid Prevalence
- Paper Alert: Preclinical Alzheimer’s Stages Predict Progression
Paper Citations
- Jack CR Jr, Therneau TM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, Mielke MM, Vemuri P, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol. 2016 Jan;15(1):56-64. Epub 2015 Nov 18 PubMed.
- Insel PS, Donohue MC, Mackin RS, Aisen PS, Hansson O, Weiner MW, Mattsson N, Alzheimer's Disease Neuroimaging Initiative. Cognitive and functional changes associated with Aβ pathology and the progression to mild cognitive impairment. Neurobiol Aging. 2016 Dec;48:172-181. Epub 2016 Aug 26 PubMed.
- Schreiber S, Schreiber F, Lockhart SN, Horng A, Bejanin A, Landau SM, Jagust WJ, Alzheimer’s Disease Neuroimaging Initiative. Alzheimer Disease Signature Neurodegeneration and APOE Genotype in Mild Cognitive Impairment With Suspected Non-Alzheimer Disease Pathophysiology. JAMA Neurol. 2017 Jun 1;74(6):650-659. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Donohue MC, Sperling RA, Petersen R, Sun CK, Weiner MW, Aisen PS, Alzheimer’s Disease Neuroimaging Initiative. Association Between Elevated Brain Amyloid and Subsequent Cognitive Decline Among Cognitively Normal Persons. JAMA. 2017 Jun 13;317(22):2305-2316. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Gothenburg
This study corroborates the strong association of APOE ε4 with amyloid buildup in the brain. Already at baseline the amyloid-positive group shows signs of neuronal impairment in the form of increased CSF concentrations of both total- and phospho-tau. Although cognitively similar at baseline, the follow-up data clearly show functional neuronal impairment in the amyloid-positive group; those group members do worse at cognitive testing and get more dementia drugs prescribed during followup.
Although the study design does not allow for drawing conclusions on causality, the data show that amyloid positivity is very common and should not to be neglected. Finally, I think it is fair to state that the study shows we have tools to detect the group that might benefit from amyloid-targeting treatments, which will be clinically important once such drugs are available.
Commonwealth Scientific and Industrial Research Organisation
Donohue and colleagues present an interesting study on the longitudinal clinical and cognitive profiles of participants with and without an abnormal burden of neocortical Aβ-amyloid. It appears that differences in cognitive testing between those with and without abnormal neocortical Aβ-amyloid burden can be detected four years after baseline examination. The clinical significance of the findings is not clear, as stated by Donohue et al. This suggests that, while baseline abnormal neocortical amyloid burden is in itself indicative of clinical and cognitive decline, it may need to be augmented by other measures of pathological burden, i.e., tau and neurodegeneration (Burnham et al., 2016; Vos et al., 2013), to elicit clinically significant decline.
The earlier differentiation between groups with and without abnormal neocortical amyloid burden on PACC, MMSE, and the CDR Sum of Boxes over the Logical Memory Delayed Recall may indicate that clinical, rather than episodic memory, measures may be more sensitive to detecting decline in preclinical populations, also identified in our paper on prodromal AD (Burnham et al., 2015). Larger studies with longer follow-up would be required to validate this.
Another interesting observation is the similar profiles of the biomarker data, regardless of high or low neocortical amyloid burden. This suggests that decline on clinical and cognitive measures is associated with abnormal pathology but not necessarily increasing pathology. The authors suggest that the presence of an APOEε4 allele is associated with faster cognitive decline; however, I wonder if the presence of an APOEε4 allele is actually associated with the baseline level of pathology (within high and low groupings) which in turn is associated with rates cognitive decline.
References:
Burnham SC, Bourgeat P, Doré V, Savage G, Brown B, Laws S, Maruff P, Salvado O, Ames D, Martins RN, Masters CL, Rowe CC, Villemagne VL, AIBL Research Group. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer's disease pathophysiology (SNAP) or Alzheimer's disease pathology: a longitudinal study. Lancet Neurol. 2016 Sep;15(10):1044-53. Epub 2016 Jul 20 PubMed.
Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM, Fagan AM. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013 Oct;12(10):957-65. PubMed.
Burnham SC, Raghavan N, Wilson W, Baker D, Ropacki MT, Novak G, Ames D, Ellis K, Martins RN, Maruff P, Masters CL, Romano G, Rowe CC, Savage G, Macaulay SL, Narayan VA, Alzheimer’s Disease Neuroimaging Initiative, AIBL Research Group. Novel Statistically-Derived Composite Measures for Assessing the Efficacy of Disease-Modifying Therapies in Prodromal Alzheimer's Disease Trials: An AIBL Study. J Alzheimers Dis. 2015;46(4):1079-89. PubMed.
Make a Comment
To make a comment you must login or register.