Does Neural Activity Drive DNA Repair?
Quick Links
Unlike most cell types, which continuously divide, neurons have to last for the life of the organism. How do they do that? One way neurons stay healthy is by repairing their DNA as they transcribe it. That is the upshot of a paper in the February 15 Nature by Michael Greenberg at Harvard Medical School and colleagues. The researchers found that the neuronal transcription factor NPAS4 doubles as a recruiter of the DNA repair complex NuA4.
- In active neurons, the transcription factor NPAS4 drives gene expression.
- It also recruits repair machinery to stitch up double-strand DNA breaks.
- Without it, damage in DNA piles up with age.
Importantly, NPAS4 binds promoters and enhancers of genes that are activated by synaptic transmission. These DNA regions are prone to double-strand breaks, the kind NuA4 can repair. In mice, knocking out NPAS4 allowed DNA damage to accumulate. It also halved their lifespan. Because all these mouse molecules are conserved in the human brain, Greenberg believes the same processes likely occur there.
Other researchers were intrigued by the implications for disease. “The results of the study … point to a potential link with neurodegenerative disorders, for which aging is the strongest risk factor,” Ekaterina Rogaeva at the University of Toronto wrote to Alzforum. Bruce Yankner, also at Harvard, agreed. “It would be of interest to explore whether genetic variants in this [NPAS4-NuA4] pathway predispose to age-related neurodegeneration,” he suggested (full comments below).
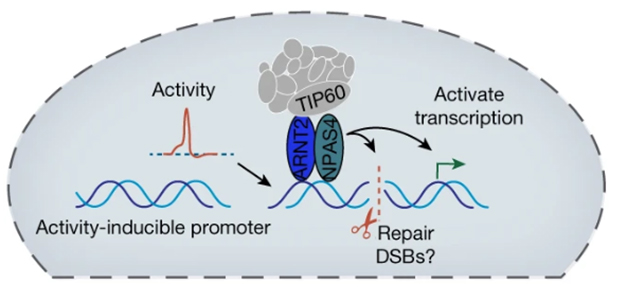
Cleanup Crew. NPAS4 (green) and its binding partner ARNT2 (blue) help recruit the NuA4 repair complex (gray) to genes activated by synaptic transmission, cleaning up DNA damage caused by transcription. [Courtesy of Pollina et al., Nature.]
Previously, Greenberg and others had identified NPAS4 as a gene switched on by neural activity that activates other genes and promotes memory formation (Lin et al., 2008; Jan 2012 news).
To learn more about how NPAS4 affects neurons, joint first authors Elizabeth Pollina and Daniel Gilliam injected kainic acid into the abdomens of transgenic mice that expressed labeled NPAS4. This induced seizures, forcing NPAS4 expression in the brain. The authors purified the labeled transcription factor from the mice’s hippocampi and used mass spectrometry to identify proteins bound to it. They discovered the NuA4 complex, which is known to facilitate repair of dsDNA breaks (Ikura et al., 2000; Sun et al., 2009). This is especially important in neurons, because they cannot clean up their DNA during replication like dividing cells can.
So far so good, but was NPAS4, in fact, essential for DNA repair? The authors mapped the chromosomal sites where NPAS4 bound, finding it at the promoters and enhancers of highly inducible genes. As expected, when neurons were electrically stimulated, double-strand breaks spiked at these sites. Neuronal activity comes with this type of DNA damage, perhaps because the breaks help untangle DNA-RNA loops that form at active transcription sites (Suberbielle et al., 2013; Madabhushi et al., 2015; Delint-Ramirez et al., 2022). By 10 hours after stimulation, most of these breaks had been repaired. However, when the authors switched off NPAS4 using a conditional knockout, the damage lingered, with about 50 percent more dsDNA breaks remaining at 10 hours than in wild-type mice.

Repair Stalled. Double-strand DNA breaks (y axis) spike two hours after synaptic stimulation. In neurons containing NPAS4 (gray), these are largely fixed soon after but in conditional NPAS4 knockout neurons (brown), strands stay broken. [Courtesy of Pollina et al., Nature.]
"The surprise was that NPAS4-NUA4 dually induced transcription in response to neuronal stimulation, and simultaneously protected those sites from DNA damage," Cynthia McMurray at Lawrence Berkeley National Laboratory, California, wrote to Alzforum. "This work provides a compelling mechanism by which cells work out an efficient way to maintain transcript stimulation and integrity during synaptic activity."
Since DNA damage accrues with age, the authors wondered if NPAS4 might counteract this in neurons. They isolated neuronal nuclei from the hippocampi of 3-month-old, 12-month-old, and 24-month-old wild-type mice. Promoter regions that do not bind NPAS4 accumulated dsDNA breaks, with old animals having about twice as many as did the young ones. At NPAS4 sites, however, the amount of DNA damage stayed constant across the lifespan, suggesting that these sites were protected.
This may affect overall longevity as well, since the NPAS4 knockout mice died at about 1 year old, half the normal lifespan. Greenberg noted that this could be due to accelerated aging, or some other consequence of NPAS4 depletion, such as excessive seizures.
Might this DNA repair mechanism go awry in neurodegenerative disease? Prior research hints at this possibility. For example, neuronal activity turns on amyloid precursor protein expression, and in different studies, either full-length APP or its AICD fragment binds to the Tip60 component of the NuA4 complex to promote DNA repair (Hass and Yankner, 2005; Mar 2009 news). These data suggest that high-octane cleavage of APP, as occurs in some familial Alzheimer’s disease cases, might interfere. “An intriguing possibility is that APP, which is integral to the pathology and genetics of Alzheimer’s disease, might modulate the activity or function of the NPAS4-NuA4-Tip60 complex,” Yankner said.—Madolyn Bowman Rogers
References
News Citations
Paper Citations
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008 Oct 30;455(7217):1198-204. Epub 2008 Sep 24 PubMed.
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000 Aug 18;102(4):463-73. PubMed.
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009 Nov;11(11):1376-82. Epub 2009 Sep 27 PubMed.
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci. 2013 May;16(5):613-21. PubMed.
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, Stott RT, Gjoneska E, Nott A, Cho S, Kellis M, Tsai LH. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015 Jun 18;161(7):1592-605. Epub 2015 Jun 4 PubMed.
- Delint-Ramirez I, Konada L, Heady L, Rueda R, Jacome AS, Marlin E, Marchioni C, Segev A, Kritskiy O, Yamakawa S, Reiter AH, Tsai LH, Madabhushi R. Calcineurin dephosphorylates topoisomerase IIβ and regulates the formation of neuronal-activity-induced DNA breaks. Mol Cell. 2022 Oct 20;82(20):3794-3809.e8. Epub 2022 Oct 6 PubMed.
- Hass MR, Yankner BA. A {gamma}-secretase-independent mechanism of signal transduction by the amyloid precursor protein. J Biol Chem. 2005 Nov 4;280(44):36895-904. PubMed.
Further Reading
News
- Somatic Mutations Accrue in Alzheimer's Neurons
- Islands of Mutated Neurons Dot the Brain. Are They Bad for Us?
- Could Genetic Mosaicism in Adult Neurons Precipitate Disease?
- Faulty DNA Repair Gene Leads to Cognitive Problems
- A Growing Problem: Oxidative Damage Drives Trinucleotide Expansions in Aging Brain
- After 40, DNA Damage Accrues in Genes, Hampering Expression
- Overworked HDACs Leave Transcriptional Posts to Push DNA Repair
- Breaking Away—Neurodegeneration Caused by DNA Damage
- C9ORF72 Throws a Wrench into DNA Repair Machinery
Primary Papers
- Pollina EA, Gilliam DT, Landau AT, Lin C, Pajarillo N, Davis CP, Harmin DA, Yap EL, Vogel IR, Griffith EC, Nagy MA, Ling E, Duffy EE, Sabatini BL, Weitz CJ, Greenberg ME. A NPAS4-NuA4 complex couples synaptic activity to DNA repair. Nature. 2023 Feb;614(7949):732-741. Epub 2023 Feb 15 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Toronto
This study identified a DNA repair mechanism involving the NPAS4–NuA4 complex, which is specific for activated neurons. This fundamental study covers function of long-lived neurons from multiple angles and is important for understanding the factors responsible for genome integrity during aging. It includes the observation that NPAS4-NuA4-bound sites are relatively protected against the accumulation of somatic mutations. The results of the study contribute to a better understanding of mechanisms of aging and point to a potential link with neurodegenerative disorders, for which aging is the strongest risk factor.
It would be important to evaluate age-related expression and the epigenetic pattern of the genes encoding the NPAS4–NuA4 complex. For instance, the number of age predictors based on DNA methylation profile (reflecting biological aging) is rising due to their potential in predicting healthspan (Bergsma and Rogaeva, 2020). This is of note because EP400, a component of NuA4, is part of the epigenetic clock reported by Zhang et al. (2019). Several recent studies of neurodegenerative diseases revealed a link between clinical outcomes and acceleration of epigenetic clocks (Bergsma and Rogaeva, 2020; Tang et al., 2022).
References:
Bergsma T, Rogaeva E. DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan. Neurosci Insights. 2020;15:2633105520942221. Epub 2020 Jul 21 PubMed.
Zhang Q, Vallerga CL, Walker RM, Lin T, Henders AK, Montgomery GW, He J, Fan D, Fowdar J, Kennedy M, Pitcher T, Pearson J, Halliday G, Kwok JB, Hickie I, Lewis S, Anderson T, Silburn PA, Mellick GD, Harris SE, Redmond P, Murray AD, Porteous DJ, Haley CS, Evans KL, McIntosh AM, Yang J, Gratten J, Marioni RE, Wray NR, Deary IJ, McRae AF, Visscher PM. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019 Aug 23;11(1):54. PubMed.
Tang X, Gonzalez-Latapi P, Marras C, Visanji NP, Yang W, Sato C, Lang AE, Rogaeva E, Zhang M. Epigenetic Clock Acceleration Is Linked to Age at Onset of Parkinson's Disease. Mov Disord. 2022 Sep;37(9):1831-1840. Epub 2022 Aug 3 PubMed.
Harvard Medical School
This elegant, multidisciplinary paper identifies a new activity-dependent NPAS4-NuA4-Tip60 complex that modulates neural circuitry and gene expression, and maintains genome stability. The findings add a new dimension to our understanding of genome maintenance in neurons, and raise interesting questions about how a breakdown in this process might affect neural circuits underlying cognition and the pathogenesis of age-related neurodegenerative disorders. The new findings advance previous observations that transient DNA damage, typically mediated by topoisomerase, is a component of transcription initiation. This process is fraught in postmitotic neurons that do not have the advantage of homologous recombination to mop up unrepaired DNA breaks. DNA damage in postmitotic neurons has been thought to be mediated primarily by non-homologous end joining (NHEJ); it will be interesting to explore the relationship of the new complex to the NHEJ machinery, and whether similar complexes, not necessarily containing NPAS4, participate in transcription-coupled repair in non-neuronal cells.
NHEJ is an error-prone mechanism of DNA repair that may contribute to the accumulation of mutations with age in postmitotic cells. In this regard, a higher rate of mutations was observed at NPAS4-bound sites relative to non-bound sites in young animals, raising the possibility that repair by this complex is also error-prone. Despite this, the mutational frequency at NPAS-4-bound sites appears to decline significantly with age (Fig. 5e). Could this reflect a contraction in NPAS-4 binding sites with age, a change in neural circuit firing patterns, or a change in the biochemistry of DNA repair in aging neurons? Whatever the explanation, the role of the complex in genome integrity and gene regulation during aging is likely to be complex.
Might this complex play a role in age-related degeneration in the brain? Interestingly, we have previously demonstrated that the amyloid precursor protein (APP) can signal to the nucleus via TIP60, an integral component of the newly identified NPAS4-NuA4-Tip60 complex (Hass and Yankner, 2005). This signaling pathway was mediated by intact transmembrane APP rather than γ-secretase cleavage of APP to Aβ, and predicted that APP cleavage might actually interfere with TIP60 function. An intriguing possibility is that APP, which is integral to the pathology and genetics of Alzheimer’s disease, might modulate the activity or function of the NPAS4-NuA4-Tip60 complex.
The role of genome integrity in Alzheimer’s disease and other age-related neurodegenerative disorders is unresolved. It may be informative, in this regard, to consider studies of humans exposed to high doses of radiation. Long-term studies of the Hiroshima atomic bomb survivors show that they are not associated with accelerated age-related cognitive decline or dementia, regardless of the age of the patient at the time of radiation exposure (Yamada et al., 2009, 2016). Moreover, patients who receive brain X-ray therapy for cancer can develop cognitive deficits, but this differs from Alzheimer’s disease both clinically and pathologically.
It is possible, however, that single, high-dose exposure to radiation is mechanistically different in terms of DNA repair than a continuous breakdown in genome stability with age. The authors note that several components of the NPAS4-NuA4 complex are mutated in neurodevelopmental disorders. It would be of interest to explore whether genetic variants in this pathway predispose to age-related neurodegeneration.
References:
Hass MR, Yankner BA. A {gamma}-secretase-independent mechanism of signal transduction by the amyloid precursor protein. J Biol Chem. 2005 Nov 4;280(44):36895-904. PubMed.
Yamada M, Kasagi F, Mimori Y, Miyachi T, Ohshita T, Sasaki H. Incidence of dementia among atomic-bomb survivors--Radiation Effects Research Foundation Adult Health Study. J Neurol Sci. 2009 Jun 15;281(1-2):11-4. PubMed.
Yamada M, Landes RD, Mimori Y, Nagano Y, Sasaki H. Radiation Effects on Cognitive Function Among Atomic Bomb Survivors Exposed at or After Adolescence. Am J Med. 2016 Jun;129(6):586-91. Epub 2015 Oct 22 PubMed.
University of California
Early studies revealed that etoposide-induced DSBs stimulated transcription of a program of immediate early genes such as FOS. DNA DSBs were subsequently observed in normal mice stimulated with fear or light in the same genes. Although DSBs were classically associated with toxicity and disease states such Alzheimer’s, collectively, these seminal studies established that DSBs could occur in the brains of normal animals. Pollina et al. now provide powerful evidence that a neuron-specific NPAS4-NUA4 chromatin complex couples synaptic activity-dependent transcription with DNA damage and repair at stimulated transcripts. The analysis is comprehensive. Knock-in mice expressing HA- and FLAG-tagged NPAS4 and NUA4 allow direct visualization by immunofluorescence, indicating that the two proteins co-localize in neurons. A physical complex between NPAS4 and NUA4 was established in vitro using IP, column chromatography, and mass spectrometry, and in vivo at genomic sites identified by cut-and-run analysis and chromatin-IP.
These convincing studies suggested that NPAS4-NUA4 assembles on chromatin in neurons in an activity-dependent manner. Although the NPAS4-NUA4 complex in other systems is known to stimulate transcription and to play a role in DNA repair, the surprise was that NPAS4-NUA4 dually induced transcription in response to neuronal stimulation and simultaneously protected those sites from DNA damage. A robust set of mapping approaches, including sBLISS and ENDSeq, confirmed that DSBs formed downstream of stimulation in genes within their promoters.
In general, the set of studies from Pollina et al. provide a tour de force of technical data to support the idea that synaptic stimulation of key transcripts and their protection from damage are linked functions. A protection system seems logical. Neurons must survive for years and must retain the integrity for synapse activation and the requisite stimulation of essential gene programs without damage. Indeed, this duality appears to be an essential process since the authors show that disruption of the NPAS4-NUA4 complex reduces lifespan.
There are reasonable gaps and additional questions raised in the analyses. For example, the authors demonstrate a dual role of NPAS4-NUA4 for a specific group of stimulated transcripts, which were preselected for harboring NPAS4-NUA4 binding sites. Since not all genes contain NPAS4-NUA4 binding sites, Pollina et al. may have opened the door to a general coupling mechanism that has shared properties with other yet-to-be characterized chromatin complexes and may influence a distinct set of transcripts that are stimulated by different conditions. Minimally, the function-protection cycle provides a general model by which activity is accompanied with a built-in protection mechanism.
While the authors clearly show the activity-dependent stimulation and repair functions of NPAS4-NUA4 were neuronal functions, it remains to be seen whether there are coupled events in astrocytes, which were not examined in these analyses. Astrocytes are intimately involved in calcium signaling. It may be that stimulation has an impact on transcript programs in astrocytes that have different kinetics and/or regulate neuronal stimulation but are masked in this study. The mechanisms by which DSBs form remains to be elucidated. There are multiple DNA DSBR complexes in these brains, and which ones are present and their machinery is, as yet, poorly understood.
Collectively, however, this work provides a compelling mechanism by which cells work out an efficient way to maintain transcript stimulation and transcript integrity during synaptic activity. Disease toxicity in Alzheimer’s may arise from interference of these protections.
Drexel University
Very exciting insights into activity-dependent DNA repair. Interestingly, we have recently discovered that, in addition to chromatin-associated mechanisms, Tip60 (KAT5) also doubles as an RNA splicing modulator. In our paper published in the Journal of Neuroscience, we propose a switching mechanism for Tip60 that allows it to regulate histone acetylation as well as splicing modulation of a similar set of Alzheimer's disease-enriched gene targets to control not only which genes are activated, but also how they are ultimately spliced for appropriate protein function.
Additionally, we show that loss of Tip60 in Alzheimer's disease model brain interferes with the splicing modulation function and may underlie some of the splicing defects detected in the postmortem Alzheimer’s patient brains. I speculate that Tip60 supports both DNA repair and splicing modulation at the sites of transcription, and that these processes go awry in the early stages of disease progression.
References:
Bhatnagar A, Krick K, Karisetty BC, Armour EM, Heller EA, Elefant F. Tip60's Novel RNA-Binding Function Modulates Alternative Splicing of Pre-mRNA Targets Implicated in Alzheimer's Disease. J Neurosci. 2023 Mar 29;43(13):2398-2423. Epub 2023 Feb 27 PubMed.
Make a Comment
To make a comment you must login or register.