Is There No End to Tau’s Toxic Tricks?
Quick Links
Besides the protein aggregates that mark tauopathies, in turns out that mutant forms of tau damage neurons in myriad other ways. Many were scrutinized at the Society for Neuroscience conference, held November 11–15 in Washington, D.C. Scientists reported how tau slows down mitochondria, disrupts the nuclear membrane and transcription, and even tangles up whole vascular networks. The meeting showed how after decades of research into the microtubule binding protein, it continues to surprise.
Tau and Organelles
Although tau is predominantly a cytoplasmic protein, several groups have begun to home in on what mutant tau does to the nucleus. Bahareh Eftekharzadeh, from Bradley Hyman’s lab at Massachusetts General Hospital, Charlestown, wondered if tau might disrupt nucleocytoplasmic transport. After all, transport falters in cells containing aggregates of other cytoplasmic proteins, such as TDP-43 and mutant huntingtin. Eftekharzadeh tested this idea using a dextran exclusion assay. It exploits the fact that if large dextran molecules of more than 500 kDa can access the nuclei, then these organelles must be compromised.
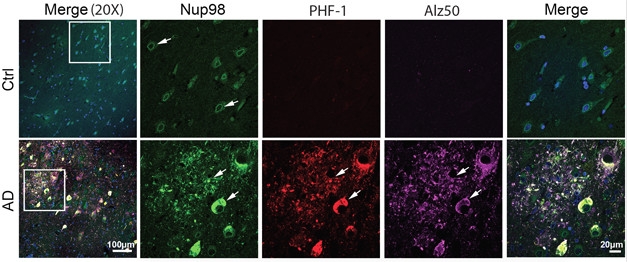
Nuclear Tau. Immunohistochemistry shows tau (PHF-1, Alz50) co-localizes with Nup98 in AD cases but not controls. [Courtesy of Bahareh Eftekharzadeh.]
To estimate the integrity of nuclei from people with AD, Eftekharzadeh tested tissue samples from the hippocampi and cerebella of patients who had died at Braak stages I, III, or VI. In all cases, nuclei from the cerebellum appeared to be intact, allowing only small dextrans of around 25 kDa to pass. By comparison, nuclei from hippocampi at Braak stage I appeared intact, but 30 percent of nuclei isolated from stage III and 50 percent of those isolated from stage VI tissue were leaky.
Why was that? Eftekharzadeh found that considerably more tau bound to nuclear pore complex proteins in the hippocampi of AD than control brains. In fact, in AD tissue about 60 percent of phospho-tau co-localized with nucleoporin 98 (Nup98). Turning to the Tg4510 mouse model of tauopathy to investigate further, she found tight co-localization of Nup98 and phospho-tau in neurofibrillary tangles. These animals express human tau with the P301L mutation and accumulate tangles in the cortex and hippocampus by four months. Almost all tangles in sections from human brain tested positive for Nup98 as well.
Eftekharzadeh thinks Nup98 might promote the formation of tangles and compromise the nuclear pore complex in the process. In support of this, she showed that, in vitro, the nucleoporin promoted aggregation of P301L tau. She needed half as much Nup98 to aggregate mutant tau as she did for wild-type tau. Surface plasmon resonance confirmed that the two proteins directly bound. Intriguingly, Eftekharzadeh said her findings support the idea that liquid-liquid phase separation might drive formation of tau tangles, because both Nup98 and tau, as Susanne Wegmann of Hyman’s lab and others reported earlier this year, form liquid droplets (May 2017 conference news; Jul 2017 news; Aug 2017 news). Fusion of the two types of droplets might set the stage for aggregation, she thinks.
In a related study, collaborator Gavin Daigle from Jeff Rothstein’s group at Johns Hopkins University, Baltimore, reported that tau binds to nucleoporins in postmortem tissue from frontotemporal dementia patients as well, and that this interferes with nucleocytoplasmic transport. Along the same theme, Garrett Lee Cornelison from Bess Frost’s lab at the University of Texas Health, San Antonio, reported that nuclear envelope invaginations in tauopathy models are enriched for polyadenylated RNA.
Invaginations and other aberrations in the nuclear envelope and nucleoskeleton have been studied before. They are a hallmark of laminopathies, where the lamin proteins that make up the nucleoskeleton fail. Since the lamin nucleoskeleton provides a scaffold for packaging DNA into highly condensed heterochromatin, transcription is altered in laminopathies.

Nuclear Invaginations. In nuclei of fruit flies that express R406W tau, lamin staining (red) outlines nuclear invaginations (left) that accumulate polyA RNA (green, center). DAPI (blue) outlines nuclei (right). [Courtesy of Garrett Cornelison.]
What does this have to do with tau? Last year, Frost reported that lamin nucleoskeleton breaks down in fly models of tauopathy, causing invagination of the nuclear envelope and neurodegeneration. She also found lamin network disruptions in tissue samples from AD patients (Frost et al., 2016). In D.C., Cornelison showed that nuclear membrane invaginations disrupt RNA processing in a tau model. He noted that these invaginations always contain nuclear pores, altering cellular homeostasis. “Diffusion from sites of transcription to nuclear pores is the rate-limiting step for RNA export,” he emphasized. Having pores in close proximity to sites of RNA synthesis, such as the nucleolus, might accelerate release of RNAs into the cytoplasm, and that could have dire consequences for the cell.
Cornelison tested this idea by blocking RNA translocation into the cytosol. The upshot: Either genetically or chemically suppressing RNA export from the nucleus protected the flies against tau toxicity. Cornelison crossed fruit flies expressing mutant tau with flies lacking Nxt1 and Nxf1, two components of the RNA export machinery. He also treated the tau flies with Leptomycin B or KPT-350, two molecules that banjax exportin 1, which ushers RNA and protein out of the nucleus. All treatments protected against tau-mediated neurodegeneration.
Further, Cornelison thinks that the nucleoplasmic reticulum actively promotes RNA export. In the R406W tau flies, he found that 43 percent of nuclei that had invaginations had more polyadenylated RNA. In fact, 32 percent of nuclear invaginations had puncta indicative of RNA accumulation in the invagination itself. Cornelison said that polyA RNA associated with nuclear invaginations in AD patients as well. He plans to investigate if particular RNAs may be involved.
Others thought these findings interesting. Eckhard Mandelkow, DZNE, Bonn, Germany, asked whether invaginations might be stabilized by tau binding to actin filaments, and what happens to invaginations if there is no tau. Cornelison said he did not know, but agreed this was an important question. He noted that nuclear invaginations happen in non-neuronal cells, particularly in cancers, which would suggest tau is not required. Mandelkow also wondered if the concentration of tau and RNA in the tiny space of the invagination might create an environment for tau to aggregate and form tangles. That would be in keeping with Eftekharzadeh’s data that Nup98 and tau co-aggregate. Frost told Alzforum that it might be the direct interaction between tau and pores that brings tau into the invaginations. Cornelison noted that flies do not seem to form the type of tangles found in the human brain, but agreed it would be interesting to test for aggregation near these invaginations.
“Collectively our studies point to the convergence of multiple tau toxicity mechanisms occurring at arguably one of the most important cellular organelles,” Eftekharzadeh told Alzforum. She noted growing evidence of pathological tau interfering with the nuclear membrane, including work on human induced pluripotent stem cell-derived neurons being studied at Rick Livesey’s lab at the University of Cambridge, England. Frost added that the leaky nuclei seen in the Hyman lab fit with pore containing invaginations, which may be less able to restrict the flow of diffusible molecules.
Another major organelle, the mitochondria, also suffers at the hands of tau. At SfN, James Johnson, who works at Michael Ashby’s lab at the University of Bristol, England, reported that mitochondria in Tg4510 mice behave erratically from an early age. Johnson infected young mice with a virus encoding a green fluorescent mitochondrial and a red fluorescent neuronal marker and repeatedly tracked both with a two-photon microscope over a cranial window. He first took measurements when the mice were 15 weeks old, and then every two weeks until week 37.
Johnson showed that while about 15 percent of neuronal mitochondria moved along axons in control mice, only 10 percent did in tau mice. As the tau mice aged, more of their mitochondria paused. They paused for longer than in normal mice, and even the mitochondria that did move were only half as fast as those from wild-type.
Whether these sluggish mitochondria explain behavioral deficits in Tg4510 mice remains to be seen. However, last March Ashby reported that dendritic spines in these animals turned over faster than those in control mice, while presynaptic spines were more stable (Jackson et al., 2017). In keeping with this, Johnson found that mitochondria in Tg4510 mice were more attracted to terminal boutons at the ends of axons and less attracted to “en passant” boutons along the axon shaft. This penchant might explain the stability of the presynapses.
Beyond Organelles
Mutant tau seems to cause problems at the tissue level, as well. Rachel Bennett, also from Hyman’s lab, reported disruptions to the vasculature in Tg4510 mice. She used two-photon microscopy to examine capillaries, showing how at 15 months, these mice have about 22,000 capillaries per cubic millimeter to 16,000 in wild-type mice. The narrowest vessels accounted for most of the difference, with the transgenic animals having many more 3 mm diameter and smaller capillaries than the wild-type. Further, Bennett reported that the small vessels twist strangely, and have nearly four times as many static white blood cells than regular vessels and very little blood flow.

Vascular Dystrophy.
Two-photon microscopy of 15-month-old mouse brain shows blood vessel abnormalities in Tg4510 mice (right) compared with wild-type controls (left). [Courtesy of Rachel Bennett.]
What causes these morphological changes? Bennett surmised that either old vessels were collapsing or that new, aberrant ones were forming. To test for the latter, she looked for expression of hypoxia-induced angiogenesis genes in endothelial cells and microglia. She found many that were more highly expressed in 15-month-old transgenics, including Pgf, Plau, Met, Serpine1, VegfA, and MMP9. Looking in mice of different ages, Bennett found that in capillaries, plasminogen activation inhibitor 1 (PAI-1), which is encoded by the Serpine1 gene, began to tick up when the animals were nine months old. It reached maximum levels when they were a year old, just before their cerebrovasculature began to morph. PAI-1 expression co-localized with cells expressing Iba1 and binding the tau aggregate antibody, MC1, hinting that microglial tau might play a role in the vasculature changes in these mice, and maybe even in some human tauopathies.
Support for this came from labmate Sarah Hopp, who outlined how microglia process tau from other cells and then release tau seeds. Hopp isolated microglia from Tg4510 mice and from APP/PS1/rTg21221 animals, which express wild-type human tau. She used quantitative PCR to measure gene expression, a Simoa assay to measure minute amounts of tau, and a cell-based assay to test for the presence of tau seeds.
Hopp found that while microglia from the tau transgenics do not express the tau gene, they do contain seeds that aggregate tau in the in vitro cell assay, indicating that the cells must have phagocytosed the protein. Even the conditioned medium from cultured microglia bore seeds, indicating that the cells then release some of the tau they consumed. Seeding activity in the medium decreased over time, suggesting a continual process of absorption and release of tau; however, it did not completely disappear, leading Hopp to speculate that the microglia cannot totally degrade the protein.
What about human microglia? Hopp isolated these cells from tissue taken from people who had just died at Braak stage VI AD or FTLD. She digested the tissue with enzymes to free the microglia, then cultured them for three days. They also harbored tau seeds and released them into the culture medium. Finally, she showed that microglia isolated from wild-type mice reduce but do not completely eliminate seeding activity in extracts from Tg4510 brain.
Hopp concluded that microglia are able to take up and break down fibrillogenic tau, but not efficiently enough to fully abolish it. She acknowledged that many questions remain open, including how the cells degrade tau, how they release it, and to what extent they contribute to the transmission of pathological tau in vivo.
Others wondered whether astrocytes might also take up and release the protein. Hopp agreed that astrocytic tau is important to understand, especially for some tauopathies, such as corticobasal dementia, where tau pathology has been documented in astrocytes. She said she has not seen tau co-localize with astrocytes, but noted that they are more difficult to isolate than microglia. On whether microglia release tau in exosomes, Hopp said it was hard to separate free from exosomal tau, though she might address the question using an ELISA with and without detergent. In response to concerns about contamination from macrophages, Hopp said she will address this by identifying cells with Tmem119, recently reported to be a microglial-specific marker (Satoh et al., 2016).
Research from many labs has indicated that while tau predominantly resides inside the cell, it can also spread trans-synaptically from neuron to neuron (Feb 2012 news). In D.C., Emily Miyoshi from Karen Gylys’ lab at the University of California, Los Angeles, proposed that this process might be spurred by Aβ. Evidence for this came from the study of human synaptosomes. Like intact neurons, these cell-free preparations respond when depolarized with potassium chloride. Previous work from Gylys’ lab showed that this caused the synaptosomes to release a C-terminal truncated form of tau (Sokolow et al., 2015).
In her poster, Miyoshi showed that the synaptosomes release tau and small vesicles, and the synaptosome medium tests positive for exosomal markers including CD9, CD63, and CD81. She then tested synaptosome extracts in the same cell-based seeding assay Hopp used. Miyoshi reported that synaptosomes from AD tissue carried almost tenfold the tau seeds as those from control samples. Furthermore, synaptosomes from two other tauopathy cases, who were at the same Braak stage as the AD patients, had no more tau seeds than control synaptosomes. Miyoshi thinks that the Aβ in the AD brain somehow makes the synaptic transmission of toxic tau more likely.
Miyoshi also claimed to have molecules that block propagation of tau. She tested this by collecting exosomes from cells that had been seeded with tau, and incubating them with naïve HEK293T cells. Two inhibitors, dubbed DDL111 and DDL112, blocked this propagation. Miyoshi declined to reveal much about these molecules beyond saying that they block the release of exosomes.—Tom Fagan
References
Research Models Citations
News Citations
- Protein Liquid-Liquid Phase Transitions: The Science Is About to Gel
- Tau Hooks Up with RNA to Form Droplets
- More Droplets of Tau
- Mice Tell Tale of Tau Transmission, Alzheimer’s Progression
Paper Citations
- Frost B, Bardai FH, Feany MB. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr Biol. 2016 Jan 11;26(1):129-36. Epub 2015 Dec 24 PubMed.
- Jackson JS, Witton J, Johnson JD, Ahmed Z, Ward M, Randall AD, Hutton ML, Isaac JT, O'Neill MJ, Ashby MC. Altered Synapse Stability in the Early Stages of Tauopathy. Cell Rep. 2017 Mar 28;18(13):3063-3068. PubMed.
- Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology. 2016 Feb;36(1):39-49. Epub 2015 Aug 6 PubMed.
- Sokolow S, Henkins KM, Bilousova T, Gonzalez B, Vinters HV, Miller CA, Cornwell L, Poon WW, Gylys KH. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. J Neurochem. 2015 May;133(3):368-79. Epub 2015 Jan 13 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.