Meta-Analysis of RNA-Seq Data is Robust, Finds Key Changes in AD
Quick Links
As some scientists create huge, new, single-nucleus RNA-sequencing datasets, others devise ways to better use existing ones. At the Alzheimer’s Association International Conference held last month in San Diego, California, Evan Macosko of the Broad Institute of Harvard and MIT in Cambridge, Massachusetts, presented his work combining small databanks into a large one. Akin to doing a meta-analysis, this approach boosts statistical power, allowing Macosko to test if transcriptome changes tied to AD progression are consistent across cohorts.
- A new inhibitory neuron subtype perishes early in Alzheimer's pathogenesis.
- Transcriptional changes in other vulnerable neurons signal hyperexcitability, broken homeostasis.
- Later on, homeostatic microglia decline while disease-associated ones appear.
Macosko and colleagues first turned to a unique cohort by Ville Leinonen, Kuopio University Hospital, Finland. Leinonen and colleagues have collected prefrontal cortex biopsies taken from 700 adults with normal-pressure hydrocephalus, a condition where excess cerebrospinal fluid swells the brain ventricles. It is treated by placing a drainage shunt into the ventricles, and this procedure allows scientists to take a tissue sample during surgery. The tissue provides a snapshot into the living brain without the peri- or postmortem artifacts that can compromise RNA stability and skew transcriptional signatures.
The researchers chose samples from older participants, because some happened to have amyloid plaques and neurofibrillary tangles. By their neuropathology scores, 29 samples had neither (A-T-), 21 had plaques but no tangles (A+T-), and eight had both (A+T+). “The beauty of this dataset is that it allowed us to see cell vulnerabilities and transcriptional signals occurring at the earliest stages of amyloid accumulation in brain tissue,” Macosko wrote to Alzforum. SnRNA-Seq of these samples yielded 1.1 million cells that clustered into 92 populations.
This high-quality dataset subsequently guided cell population annotations of another 1.3 million cells from 29 published human and mouse snRNA-Seq datasets. Sixteen of those came from postmortem cortical tissue, six were from AD cohorts. This yielded a total of 2.4 million cells. To align them, the scientists used two analysis tools, LIGER, which the Macosko lab had developed, and Harmony. Both helped them cluster cells by type across these 30 datasets, by way of genetic markers (see image below; Welch et al., 2019; Korsunsky et al., 2019).
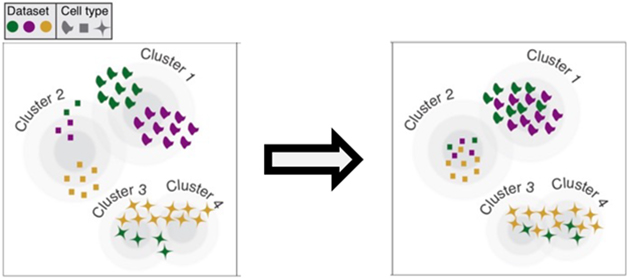
Reshuffle by Cell Type. Algorithms align cell clusters from different datasets (colors) to generate one cluster for each cell type (symbols). [Courtesy of Evan Macosko, Broad Institute.]
Which AD-linked cell changes jumped out of this combined dataset? At the earliest stage, i.e., in cases that had few amyloid plaques but no tau tangles, three neuronal populations tended to be already depleted from the cortex relative to pathology-free tissue: layer 1 inhibitory interneurons, layer 2 excitatory neurons, and layer 4 excitatory neurons. Moreover, interneurons from these amyloid-positive samples expressed fewer genes.
Notably, the excitatory neurons had been previously reported as depleted in entorhinal cortex tissue affected by AD, while the inhibitory ones had never been reported as MIA in AD, according to Macosko. Kyle Travaglini, Allen Institute for Brain Science, Seattle, said that he also saw these layer 1 interneurons die off, though interneurons in layers 2 and 3 degenerated more in his dataset. The Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) represents a new, large snRNA-Seq effort, containing 1.2 million cells from 84 people across the AD pathological spectrum (see Part 17 of this series).
In samples that had cortical tau tangles in addition to amyloid plaques, dubbed later-stage AD, microglial changes took center stage. Homeostatic CX3CR1-expressing microglia were depleted, while disease-associated microglia expressing the marker LPL were abundant. Not every individual dataset contained evidence of these DAMs because, on their own, they lacked the statistical power to detect such small cell subpopulations, Macosko said.
What about transcriptional changes across disease severity? In other words, do these disease-associated microglia and the vulnerable neurons express the same or different genes in early and later-stage samples? In this analysis, DAMs had the same differentially expressed genes (DEGs), many of which function in interferon signaling, throughout the progression of AD captured here, with expression higher in later-stage samples.
In contrast, layer 2 and 4 excitatory neurons changed. They overexpressed DEGs related to glycolysis, handling of reactive oxygen species, and synaptic vesicle processing—but only at the early stage. “This suggests that neurons are being taxed with hyperexcitability and are trying to manage and maintain homeostasis, which peters away at later stages of disease,” said Macosko. During later-stage AD when plaques and tangles are present, the same upper-layer excitatory neurons downregulate synaptic and neurotransmission genes, perhaps foretelling the impending loss of these neurons.
All told, Macosko thinks that the vanishing of specific inhibitory interneurons may cause overexcitability early in AD, eventually leading to the well-described neuron loss at later stages. Hyperactivation in early affected people has been described at other levels of investigation, for example by fMRI, but not at a single-cell transcriptional level (e.g., Dickerson and Sperling, 2008).
More analyses of these cumulative datasets are yet to come. “By incorporating more data as it comes out, we hope to make more and more insights into AD biology using this integration and analysis platform,” Macosko said.
Following SEA-AD’s lead, Macosko plans to make this dataset publicly available, together with tools that allow researchers to integrate their own snRNA-Seq data into the database.—Chelsea Weidman Burke
References
News Citations
Paper Citations
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell. 2019 Jun 13;177(7):1873-1887.e17. Epub 2019 Jun 6 PubMed.
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019 Dec;16(12):1289-1296. Epub 2019 Nov 18 PubMed.
- Dickerson BC, Sperling RA. Functional abnormalities of the medial temporal lobe memory system in mild cognitive impairment and Alzheimer's disease: insights from functional MRI studies. Neuropsychologia. 2008;46(6):1624-35. PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.