Genetics of DLB: Setting Up to Fill a Mostly Empty Canvas
Quick Links
Besides ample new data presentations, the International Dementia with Lewy Body Conference December 1-4 in Fort Lauderdale, Florida, featured a reprise of the field's perennial debate. It is the verbal tug-of-war between movement disorder and dementia specialists about whether research on the spectrum of Lewy body disorders is well served by clinical classifications such as dementia with Lewy bodies (DLB) and Parkinson’s dementia (PDD). Perhaps a different scientific approach can inform this old parley with fresh information? Indeed, what do geneticists say about DLB as a unique entity? Thus far, they know little, but several presenters showed how the question is starting to draw attention on a larger scale.
Jose Bras of University College London, U.K., reminded the audience that late-onset Alzheimer’s and Parkinson’s were not considered genetic diseases until recently. By now some 30 genes are known to be involved in each of them, and their overall heritability is estimated at 60 and 28 percent, respectively. At the level of GWAS, the known common genetic risk factors for AD and PD do not overlap (Moskvina et al., 2013). Yet there is DLB, a clinical hybrid of the two that is, at least according to its proponents, as common as it is overlooked and underserved. DLB genetics is just beginning to pick up steam, with some 100 papers in the literature compared with 1,000 on the genetics of PD and 1,800 on that of AD.
To date, scientists have implicated four genes in DLB. They are:
- APOE, which was first reported 21 years ago and subsequently confirmed (Hardy et al., 1994).
- SNCA (encoding the α-synuclein protein) by way of a clinical DLB presentation in some PD families carrying a gene triplication (Singleton et al., 2003).
- A locus on chromosome 2 in a large family (Bogaerts et al., 2007).
- GBA, variants of which drive up a person’s risk of developing DLB (Tsuang et al., 2012).
These first clues come from research with single families or samples of fewer than 100 patients.
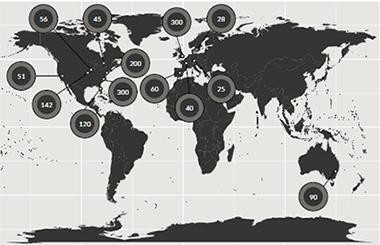
About 1,400 pathologically confirmed cases from three continents make up an ongoing GWAS of DLB. [Courtesy of Jose Bras, Rita Guerreiro, UCL.]
To inject more power into the gene hunt, Bras and collaborators in Europe, Australia, and across North America have thus far pooled 1,400 cases confirmed by neuropathology to have had DLB. This is still a far cry from the nearly 100,000 people whose samples are available for AD/PD GWAS, but it’s a first step toward a comprehensive look at the genetic signature of DLB, Bras said. This ongoing project uses the NeuroX array, a new tool geneticists including Bras designed for rapid and cost-effective genotyping in neurodegeneration research (Nalls et al., 2015).
Samples of this size support heritability estimates. These calculations put DLB’s genetic component at 31 percent, close to that of PD and half that of AD, Bras told the audience. They also enable estimates of the genetic overlap among AD, DLB, and PD—diseases already known to overlap at the clinical and the neuropathological level. Genetically speaking, DLB correlates strongly with AD, though much of that is due to ApoE. Subtracting ApoE leaves DLB equally correlated to AD and PD, Bras reported. In contrast, AD and PD in this calculation again came out to be genetically uncorrelated. This research appeared in Neurobiology of Aging online last month (see Guerreiro et al. 2015).
Last December, Bras, collaborator Rita Guerreiro of UCL, and colleagues around the world reported preliminary results from an early data freeze of 750 samples from this growing cohort (Bras et al., 2014). This initial analysis did not survey the whole genome in an unbiased manner, but rather examined 54 genomic regions previously associated with PD and AD. Extracting variants within 500 kilobases of the known top hits from AD and PD GWAS, this study reproduced APOE as the strongest signal but also showed study-wide significant signals around SNCA, the gene for α-synuclein, and SCARB2, a lysosomal gene implicated in PD.
Because the gene chip used in these GWAS only pointed to APOE without distinguishing between the ApoE2, 3, and 4 alleles, Guerreiro decided to nail down the precise nature of the link. To do that, she harked back to the early days of DNA sequencing. She isolated DNA from the DLB patient samples, digested it with a restriction enzyme, loaded the fragments onto a gel, and compared their relative sizes to deduce the APOE genotypes. This direct genotyping of 679 DLB DNA samples showed that the APOE hit in previous GWAS is indeed due to ApoE4, Guerreiro reported in Fort Lauderdale. Specifically, the frequency of the ApoE4 risk allele in the DLB cohort, at 28.1 percent, lay midway between that of the general population (13.7 percent) and that in AD (36.7 percent). Conversely, the protective ApoE2 allele in DLB, at 5.4 percent, lies midway between its frequency in the general population (8.4 percent) and people with AD (3.9 percent). ApoE4-carrying DLB patients also tended to die younger, Guerreiro reported.
As for SNCA and SCARB2, even though these genes had come up in previous PD GWAS, a deeper look at their respective genomic regions pinpointed different individual markers in both the SNCA and SCARB2 genes for DLB and for PD. In other words, even though both genes are involved in both diseases, the most significant markers appear to be in different areas of those genes in DLB and PD. “We do not know what this means functionally yet, though it may have to do with differential control of gene expression,” Bras said.
Beyond this, the researchers are carrying out a full-fledged GWAS using the newer NeuroX2 array, which is stocked with additional, lower-frequency variants to drill deeper into the genetics of DLB. Giving a status update on this ongoing work, Bras said that to date, genotyping of 1,200 pathologically proven cases has replicated the known genes and dug up additional hits he deemed too preliminary to name. In addition, ongoing exome sequencing of the entire cohort plus some familial cases thus far has generated nearly 10,000 new, high-impact variants of yet-unknown clinical significance. Some are in genes known to be involved in AD and PD, such as GBA, PSEN2, SCARB2, TREM2, GRN, TYROBP, APOE, and LRRK2, Bras showed.
In his talk, Ziv Gan-Or of McGill University in Montreal echoed the theme of the same genes showing up in overlapping diseases by way of different markers. In Fort Lauderdale, Gan-Or described an ongoing sequencing and genome-wide association study of 1,200 people with REM Sleep Behavior Disorder and as many controls. RBD is increasingly seen as a prodromal stage of DLB or PD. While this work is ongoing, Gan-Or noted that the genetics of people with this sleep disorder is emerging to be similar overall to that of people with DLB. Some of the same genes that are known from previous genetic studies of α-synucleinopathies are coming up in this RBD GWAS. In particular, several known PD- or DLB-linked mutations in GBA and SCARB2 were associated with RBD. In the future, genetics and clinical data together may be able to identify people with this sleep disorder who are likely to progress to DLB or PD within the time frame of a clinical trial (see Part 1 of this series). Data from a smaller study leading up this the current project were recently published (Gan-Or et al., 2015).
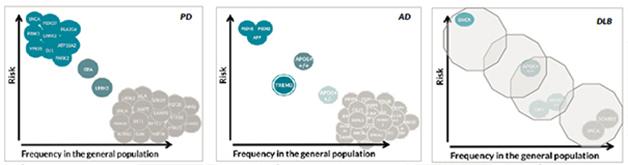
The genetics of dementia with Lewy bodies, unlike that of Alzheimer’s or Parkinson’s, is mostly unknown. Hardly any common but low-risk variants or rare, high-risk variants have been discovered. [Courtesy of Jose Bras, Rita Guerreiro, UCL.]
What does this current information imply for the genetic architecture of DLB? This question is often addressed with a diagram that depicts the risk a given variant confers over how frequently it occurs in a population. The most frequent variants confer small amounts of risk—unsurprisingly, because otherwise much of the population would have the disease. The highest risk comes from rare, autosomal-dominant mutations. For both AD and PD, dozens of low-risk variants are known, as are smaller numbers of highly penetrant variants. Some genes, such as SNCA, can show up in both corners, once as a triplication causing rare, early onset disease and once as a common variant contributing minimally to a person’s overall genetic risk. The middle space features APOE and TREM2 for AD and GBA and LRRK2 for PD, but largely awaits findings from ongoing deep-sequencing initiatives. In DLB, Bras said, this diagram remains an almost empty canvas, except for a few initial dots previously known from AD and PD. “So far we have not yet identified any genes specific for DLB. All genes we know to be involved in DLB are also involved in AD or PD. The work we are doing now will allow us to improve this,” Bras said.
“This data certainly suggests that there is something about the genetics of DLB that will be different than AD or PD,” said Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. Scientists are waiting to see if that “something” is a particular multigenic predisposition, or a gene unique to DLB, or perhaps variants that drive the disease-specific topography of a given pathogenic protein.
In Fort Lauderdale, Cyrus Zabetian of the University of Washington, Seattle, presented a comparison of candidate genes in a smaller sample of 348 neuropathologically confirmed DLB cases and 102 PDD cases from seven centers in the United States. Finding broadly similar results, this study suggested that ApoE4 is more common in DLB, the disease that is arguably closer to AD, than in PDD, the disease nearer to PD. “It was striking that homozygous carriers of ApoE4 were greatly overrepresented among DLB cases,” Zabetian said. Au contraire, pathogenic variants of GBA were more common in the PDD patients than the DLB patients, offering a genetic hint of where those genes lie along the Lewy body diseases spectrum that spans from AD to DLB to PDD to PD. “If you carry ApoE4, it pushes you toward an earlier presentation of dementia, hence you are more likely to be classified as having DLB. GBA mutations, on the other hand, push you toward α-synucleinopathy, so you are more likely to be classified as having PDD,” Zabetian said.
Another hint that ApoE4 puts its finger firmly on the AD end of the DLB scale came from a study relating ApoE genotype to neuropathology in large series of people who had had a diagnosis of DLB during life. Melissa Murray from the Mayo Clinic in Jacksonville, Florida, reported that while ApoE4 carriers tended to have worse α-synuclein pathology than non-carriers, that link was due to the presence of amyloid pathology. About 90 percent of patients with DLB have AD pathology, as well, (see Part 3 of this series). In AD research, ApoE4 is increasingly seen as an indicator that amyloid is present in the brain with age, and several presentations in Fort Lauderdale, including Murray’s, pointed in the same direction for DLB.
At the conference, some scientists voiced doubt that genetics would ever be as instructive in DLB as it has become in frontotemporal dementia, in part because few large pedigrees are known. “In the clinic, by and large DLB is not an inherited disorder,” agreed Ian McKeith of Newcastle University, U.K. At the same time, several affected people who had come for the concurrent patient and care-partner track of this conference brought up a family pattern of DLB. They asked the scientists to explain their possible genetic versus environmental risk, for example among farming families in whom adult sons and cousins are wondering if handling the same pesticides as had their affected father and uncles will predispose them to a Lewy body disorder. Asked like this, the scientists had to answer simply, “We do not know.”—Gabrielle Strobel
References
News Citations
- Dementia with Lewy Bodies: Is the Research Ready For Clinical Trials
- Dementia with Lewy Bodies: Sharper Image for a Formerly Blurry Disease
Paper Citations
- Moskvina V, Harold D, Russo G, Vedernikov A, Sharma M, Saad M, Holmans P, Bras JM, Bettella F, Keller MF, Nicolaou N, Simón-Sánchez J, Gibbs JR, Schulte C, Durr A, Guerreiro R, Hernandez D, Brice A, Stefánsson H, Majamaa K, Gasser T, Heutink P, Wood N, Martinez M, Singleton AB, Nalls MA, Hardy J, Owen MJ, O'Donovan MC, Williams J, Morris HR, Williams NM. Analysis of Genome-Wide Association Studies of Alzheimer Disease and of Parkinson Disease to Determine If These 2 Diseases Share a Common Genetic Risk. JAMA Neurol. 2013 Aug 5; PubMed.
- Hardy J, Crook R, Prihar G, Roberts G, Raghavan R, Perry R. Senile dementia of the Lewy body type has an apolipoprotein E epsilon 4 allele frequency intermediate between controls and Alzheimer's disease. Neurosci Lett. 1994 Nov 21;182(1):1-2. PubMed.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003 Oct 31;302(5646):841. PubMed.
- Bogaerts V, Engelborghs S, Kumar-Singh S, Goossens D, Pickut B, van der Zee J, Sleegers K, Peeters K, Martin JJ, Del-Favero J, Gasser T, Dickson DW, Wszolek ZK, De Deyn PP, Theuns J, Van Broeckhoven C. A novel locus for dementia with Lewy bodies: a clinically and genetically heterogeneous disorder. Brain. 2007 Sep;130(Pt 9):2277-91. PubMed.
- Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, Kramer P, Woltjer R, Kukull W, Nelson PT, Jicha GA, Neltner JH, Galasko D, Masliah E, Trojanowski JQ, Schellenberg GD, Yearout D, Huston H, Fritts-Penniman A, Mata IF, Wan JY, Edwards KL, Montine TJ, Zabetian CP. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012 Nov 6;79(19):1944-50. PubMed.
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, Saad M, Jansen I, Guerreiro R, Lubbe S, Plagnol V, Gibbs JR, Schulte C, Pankratz N, Sutherland M, Bertram L, Lill CM, DeStefano AL, Faroud T, Eriksson N, Tung JY, Edsall C, Nichols N, Brooks J, Arepalli S, Pliner H, Letson C, Heutink P, Martinez M, Gasser T, Traynor BJ, Wood N, Hardy J, Singleton AB, International Parkinson's Disease Genomics Consortium (IPDGC), Parkinson's Disease meta-analysis consortium. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging. 2015 Mar;36(3):1605.e7-12. Epub 2014 Aug 4 PubMed.
- Guerreiro R, Escott-Price V, Darwent L, Parkkinen L, Ansorge O, Hernandez DG, Nalls MA, Clark L, Honig L, Marder K, van der Flier W, Holstege H, Louwersheimer E, Lemstra A, Scheltens P, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Ortega-Cubero S, Pastor P, Ferman TJ, Graff-Radford NR, Ross OA, Barber I, Braae A, Brown K, Morgan K, Maetzler W, Berg D, Troakes C, Al-Sarraj S, Lashley T, Compta Y, Revesz T, Lees A, Cairns NJ, Halliday GM, Mann D, Pickering-Brown S, Powell J, Lunnon K, Lupton MK, International Parkinson's Disease Genomics Consortium, Dickson D, Hardy J, Singleton A, Bras J. Genome-wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson's and Alzheimer's diseases. Neurobiol Aging. 2016 Feb;38:214.e7-10. Epub 2015 Nov 2 PubMed.
- Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, Hernandez DG, Nalls MA, Clark LN, Honig LS, Marder K, Van Der Flier WM, Lemstra A, Scheltens P, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Ortega-Cubero S, Pastor P, Ferman TJ, Graff-Radford NR, Ross OA, Barber I, Braae A, Brown K, Morgan K, Maetzler W, Berg D, Troakes C, Al-Sarraj S, Lashley T, Compta Y, Revesz T, Lees A, Cairns N, Halliday GM, Mann D, Pickering-Brown S, Dickson DW, Singleton A, Hardy J. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet. 2014 Dec 1;23(23):6139-46. Epub 2014 Jun 27 PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.