Gene Therapies Enter Trials for Many Brain Pathologies—What about AD?
Quick Links
Buoyed by success in spinal muscular atrophy, scientists are once again devising gene-based therapies for neurodegeneration. Some are permanent, which makes proving their safety all the more challenging. Still, as shown at this year’s annual meeting of the Society for Neuroscience, held October 19–23 in Chicago, gene-based therapies for Alzheimer’s, Parkinson’s, frontotemporal dementia, amyotrophic lateral sclerosis, and some rare neurodevelopmental disorders are in clinical testing (see Nov 2019 conference news). Others are in preclinical development, as scientists are looking to suppress Aβ production, boost immunotherapy, or restore neurons lost to neurodegenerative disease.
Gong Chen and colleagues at Penn State University, in University Park, are pursuing the latter. “One reason so many clinical trials for AD have failed may be that too many neurons have already been lost,” Chen said. Previously, researchers in his lab reported that a single transcription factor, NeuroD1, can reprogram astrocytes into neurons, offering a potential way to replenish neurons in later stages of disease (Guo et al., 2014). This year, Chen reported that when spliced into an AAV9 vector and injected into the brain, NeuroD1 restored function after a stroke in both mice and nonhuman primates (Chen et al., 2019; Ge et al., 2019). NeuroD1 did that by creating not only new neurons, but astrocytes as well, as it prompts astrocytes to both divide and differentiate. The new astrocytes seemed to attract new blood vessels. “We are essentially regenerating new neural circuitry,” said Chen. Other groups are using similar, antisense oligonucleotide approaches to suppress transcription factors and steer progenitors toward becoming dopaminergic neurons to replenish those lost in in Parkinson’s disease (Jul 2019 conference news).
Could AAV-NeuroD1 work for AD? Chen and colleagues tried it in 5xFAD mice. “We regenerated millions of new neurons throughout the brain,” said Chen (see image below). The neurons survived at least eight months, while the number of reactive astrocytes fell. Mice treated with the vector learned and remembered better, finding a platform hidden in a water maze faster than untreated controls, Chen claimed. He is testing the vector in a nonhuman primate model of AD in China.
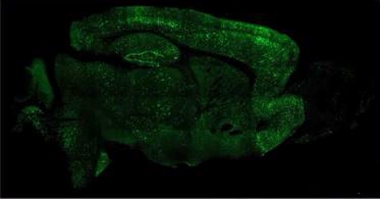
Cellular Conversion. Injected into the mouse brain, AAV-NeuroD1 generates millions of neurons (green) from astrocytes. [Courtesy of Gong Chen, Penn State University.]
Chen hopes the strategy will work in other diseases, as well. When injected into the spinal cords of mice carrying the G93A SOD1 mutation that causes ALS, AAV-NeuroD1 vectors restored motor neurons throughout the spinal cord, and improved motor function, he reported at SfN.
Others are trying to boost immunotherapies with gene-based tricks. Over the last 20 years, scientists have targeted a volley of therapeutic antibodies at various toxic proteins linked to neurodegeneration; alas, those immunoglobulins poorly penetrate the brain, usually at less than 1 percent of the injected dose. AAV offers a way of having them made right in the parenchyma, where they are needed.
One such therapy is an AAV9 carrying the genetic code for an anti-vascular endothelial growth factor antibody. Called RGX-314, it is in clinical trials for age-related macular degeneration. It was developed by REGENXBIO, Rockville, Maryland, which has partnered with Neuroimmune in Zurich to develop “vectorized antibodies” for neurodegenerative diseases.
In Chicago, REGENXBIO’s Olivier Danos said that this partnership will first target tau. His company holds patents on more than 100 novel AAV vectors, including AAV7, AAV8, AAV9, and AAVrh10, and is developing yet more variants based on tweaks to the capsid encasing the genetic information. Neuroimmune’s calling card is its strategy of screening healthy old people’s blood for naturally occurring antibodies that stave off diseases of aging. Aducanumab, a Phase 3 anti-Aβ antibody, was found that way (Oct 2019 news), as was the Phase 1 tau antibody BIIB076. Danos did not elaborate on which tau antibodies he will vectorized.
As for amyloid, Sergio Ferreira and colleagues at the Federal University of Rio de Janeiro developed Nusc1, a single-chain, variable fragment antibody whose DNA can be more easily packaged into AAV-9 than a full-size immunoglobulin. At SfN, Ferreira reported that Nusc1 binds to large Aβ oligomers in the human brain and neutralizes Aβ toxicity in cultured neurons, while having little affinity for Aβ monomers or fibrils (Sebollela et al., 2017). In vitro, hippocampal neurons infected with AAV-Nusc1 produced the antibody, which protected the cells from Aβ toxicity while control neurons bound Aβ oligomers and lost dendritic spines. When Maria Selles in Ferreira’s lab injected AAP/PS1 mice with AAV-Nusc1, it reversed memory deficits in novel-object-recognition tests. Ferreira hopes to test the therapy in clinical trials.
Other labs are starting to ramp up gene-therapy programs. “It feels really promising to be able to target the fundamental cause of a disease,” said Nick Fox, University College London. Fox believes that for some diseases, including familial AD, gene-based therapy could be the way of the future. “With single-gene disorders we have no doubt about the cause of the problem, and with these technologies we know we can go after it,” he said. Fox thinks a treatment for FAD will come before there is one for sporadic AD, which would be ironic, he said, given that people with FAD were told for years that they were not eligible for AD clinical trials.
Fox leads a gene-based research initiative at the UCL affiliate Queen Square Institute of Neurology. Funded by a £5 million donation from the Sigrid Rausing Trust, the Neurogenetics Therapies Programme plans to develop treatments for a variety of neurodegenerative diseases. “We have a strong desire to do the best for the patients and families we work with on a regular basis,” Fox said. The institute has been building cohorts of single-gene disorders such as Huntington’s, Alzheimer’s, and tauopathies since the 1980s.
“We want to create a genetic-therapy center where we can pool our expertise and resources, build a common infrastructure, share lessons learned, and formulate best practices,” said Fox. UCL’s Sarah Tabrizi and Cath Mummery have been collaborating with Ionis Pharmaceuticals, Carlsbad, California, to develop antisense oligonucleotides (ASOs) that target, and suppress expression of, mutant huntingtin and mutant tau, respectively. In Phase 1/2, IONIS-HTTRx halved the amount of mutant huntingtin in the CSF of carriers; licensed to Roche, this therapy is in Phase 3 (Mar 2018 conference news). The question with ASOs in general is whether they penetrate the parenchyma of the large human brain deeply enough to reach their targets. UCL is a site for Phase 1 testing of the tau ASO Ionis-MAPTRx.
Fox anticipates multiple preclinical projects at UCL, both AAV- and ASO-based, that build on the HD and tau programs to target AD, frontotemporal dementia, and other diseases. The UK DRI has slated £2 million for a gene-therapy program at King’s College London, where Chris Shaw will lead development of AAV-based vectors to treat ALS.
Other preclinical, gene-based therapies for AD include a project at Harvard University led by Connie Cepko to boost levels of Nrf2. This transcription factor protects neurons by turning on genes combating oxidative stress. At the University of California, San Diego, Mark Tuszynski, leads an AAV2 program to boost brain-derived neurotrophic factor production in the brain, and has completed a pilot trial in nonhuman primates (Xiong et al., 2015; Nagahara et al., 2018).
None of this is Easy
The researchers are well aware of challenges ahead. Some of the more-heterogeneous diseases, including late-onset AD, may be unsuitable. “We don’t have a good enough understanding of what would be a good target for sporadic AD,” said Bart De Strooper, U.K. Dementia Research Institute, London. Perhaps knocking down expression of APP or ApoE, but that might require suppression for five to six years, he speculated. The idea is to start with FAD and build from there.
How about regulating microglia? That might be further off, De Strooper believes. Microglia may be trickier to target via AAV than neurons, because the genetic material in AAV incorporates into an episome that stays put in the nucleus as long as the cell does not divide. That works well for postmitotic neurons; however, microglia are continually replaced, meaning the episome could be lost.
Repeat injections to replenish lost vectors are dangerous because the body mounts an immune response to the capsid the first time, hence a second injection could elicit a massive inflammatory response. “This is a major issue in the field right now,” said Petra Kaufmann, AveXis, Bannockburn, Illinois. Kaufmann said it might work to use a different vector the second time, or develop a way of first administration that allows a second, or to suppress the immune system. The latter is already being done. In trials of zolgensma, babies, who received up to 200 trillion virus particles per Kg, were also given prednisolone for 30 days.
It may be necessary to sidestep microglia to limit immune responses. “We want to minimize antigen presentation,” said Beverly Davidson, Children’s Hospital of Philadelphia. Even so, microglia might be targeted indirectly by “cross correction,” whereby a vector delivered to one cell type makes a protein that is secreted and taken up by another cell type.
Davidson has used this approach successfully to correct lipofuscinosis in dogs carrying loss-of-function mutations in the tripeptidyl peptidase 1 gene. This enzyme cleaves tripeptides from the N-terminal of proteins in the lysosome. In people, such mutations cause a form of Batten’s disease. When Davidson targeted ependymal cells with an AAV carrying the wild-type gene, the cells expressed the peptidase and released it into the CSF, whereupon it was taken up by parenchymal cells. Davidson showed videos of two 9-month-old Dachshunds with TPP1 deficiency. An untreated dog walked with extreme difficulty, tipping over every few steps, while the one treated with AAV-TPP1 ran around like a normal puppy (Katz et al., 2015). Davidson has also engineered AAV capsids to target endothelial cells, which then release the expressed protein into the lumen of the blood vessel. Of course, she noted, only proteins that are secreted by one cell type and taken up by another are suitable for cross correction.
Others warned that science needs to understand targets comprehensively before knocking them down with gene therapy. That is especially true for AAV. While antisense oligonucleotides will wash out once treatment stops, the AAV episome is permanent, at least in neurons. “This is a big risk,” agreed De Strooper. “I hope that at some point in the future we will have the means to control these vectors, maybe with promoters that are inducible and self-deactivating.” Clive Svendsen, Cedars-Sinai Regenerative Medicine Institute, Los Angeles, has used just such an approach in the engineered GDNF gene, which only turns on when doxycycline is present. “Being able to turn the gene on and off could be extremely important when this gene therapy is used in humans,” Svendsen said.
The challenges for neurodegenerative disease do not end there. Another one is getting enough of the vector into the brain and keeping it there. In Chicago, Danos showed that even when injected directly into the macaque central nervous system through the cisterna magna, AAV9 ended up in the thyroid, liver, spleen, and other organs within three days.
In macaques, AAV9 vectors caused some degeneration of dorsal root ganglia (DRG) accompanied by infiltration of immune cells, plus some axon damage in the spinal cord, though the animals’ behavior remained normal. In piglets, however, dorsal root ganglia lesions were accompanied by proprioceptive deficits and ataxia. On October 30, the FDA put a hold on STRONG, Novartis’ Phase 1 study of zolgensma in older infants, because of such animal data. And on November 12, the FDA halted, for a second time, a Phase 1/2 trial of SGT-001, a gene therapy for Duchenne muscular dystrophy developed by Solid Biosciences, Cambridge, Massachusetts. In a testament to the dangers of systemically administering AAV, this virus caused an immune response, loss of red blood cells, and kidney and lung damage in one of three patients who had received the therapy in October.
REGENXBIO, Davidson's, and many other groups are tweaking capsid proteins to improve targeting. For example, Sophie Mathiesen, University of Otago, Dunedin, New Zealand, described a modified capsid called PHP.eB. Unlike AAV9, it ignores the liver but infiltrates cells throughout the brain, at least in mice (see related Dec 2017 conference news). Still, Davison cautioned that, in some cases, pan-neurotropic viruses will not be good enough, and that they will have to be optimized to specific neuronal subtypes. Ideally, one wants to have ways to turn the viruses off, she said.
When asked about DRG side effects of his vectors, Abeliovich said that his team had specifically looked for this and found nothing. “We’ve tried multiple different doses in nonhuman primate models and looked specifically at DRG and white matter. At up to 1x1014 units/g of brain, we do not see any such effects,” he said. He thinks that the doses used in those other studies were so high that the DRGs became flooded with viral particles. “But these vectors are not all the same, and the most obvious difference is the cargo,” he said.
Danos agreed. “It could be the capsid, the expression level, toxicity linked to the CpG content of the genetic cargo … those are difficult to reproduce in different systems,” he said. “The reassuring message is that, as a rule, the vectors do not appear to be very toxic. But we need to look into it.”—Tom Fagan
References
News Citations
- Time to Try Again: Gene-Based Therapy for Neurodegeneration
- Dopaminergic Neurons Conjured from Astrocytes Restore Motion
- ‘Reports of My Death Are Greatly Exaggerated.’ Signed, Aducanumab
- Antisense Therapy Cuts Huntingtin Protein in CSF by Half
- Tau Snapshots from Neuroscience 2017
Therapeutics Citations
Paper Citations
- Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G. In Vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell. 2014 Feb 6;14(2):188-202. Epub 2013 Dec 19 PubMed.
- Chen YC, Ma NX, Pei ZF, Wu Z, Do-Monte FH, Keefe S, Yellin E, Chen MS, Yin JC, Lee G, Minier-Toribio A, Hu Y, Bai YT, Lee K, Quirk GJ, Chen G. A NeuroD1 AAV-Based Gene Therapy for Functional Brain Repair after Ischemic Injury through In Vivo Astrocyte-to-Neuron Conversion. Mol Ther. 2019 Sep 6; PubMed.
- Sebollela A, Cline EN, Popova I, Luo K, Sun X, Ahn J, Barcelos MA, Bezerra VN, Lyra E Silva NM, Patel J, Pinheiro NR, Qin LA, Kamel JM, Weng A, DiNunno N, Bebenek AM, Velasco PT, Viola KL, Lacor PN, Ferreira ST, Klein WL. A human scFv antibody that targets and neutralizes high molecular weight pathogenic amyloid-β oligomers. J Neurochem. 2017 Jul 3; PubMed.
- Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, Cepko CL. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest. 2015 Apr;125(4):1433-45. Epub 2015 Mar 23 PubMed.
- Nagahara AH, Wilson BR, Ivasyk I, Kovacs I, Rawalji S, Bringas JR, Pivirotto PJ, Sebastian WS, Samaranch L, Bankiewicz KS, Tuszynski MH. MR-guided delivery of AAV2-BDNF into the entorhinal cortex of non-human primates. Gene Ther. 2018 Apr;25(2):104-114. Epub 2018 Mar 13 PubMed.
- Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger FA, Young WM, Johnson GC, Whiting RE, Coates JR, Davidson BL. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci Transl Med. 2015 Nov 11;7(313):313ra180. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Hordeaux J, Hinderer C, Buza EL, Louboutin JP, Jahan T, Bell P, Chichester JA, Tarantal AF, Wilson JM. Safe and Sustained Expression of Human Iduronidase After Intrathecal Administration of Adeno-Associated Virus Serotype 9 in Infant Rhesus Monkeys. Hum Gene Ther. 2019 Aug;30(8):957-966. Epub 2019 Jun 10 PubMed.
- Hordeaux J, Hinderer C, Goode T, Katz N, Buza EL, Bell P, Calcedo R, Richman LK, Wilson JM. Toxicology Study of Intra-Cisterna Magna Adeno-Associated Virus 9 Expressing Human Alpha-L-Iduronidase in Rhesus Macaques. Mol Ther Methods Clin Dev. 2018 Sep 21;10:79-88. Epub 2018 Jul 14 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Rowan University, NJ Institute for Successful Aging
A great deal of preclinical work in gene therapy for diverse neurological diseases has been conducted over the past 20 years. The technologies for both antisense and viral-vector-based delivery are finally becoming mature, and without a doubt will be applied in the next few years to complex diseases such as Alzheimer's disease.
NIH and industry need to partner to support these important, pioneering clinical trials. While some transcription factors might make for good gene therapy approaches, I'm highly skeptical about approaches such as NeuroD1, which may work well in the lab but are unlikely to be the linchpin for effective therapies.
Also, growth factor-based approaches were tried in the past with human gene therapy, but are unlikely to be sufficient on their own to reverse the disease course.
Make a Comment
To make a comment you must login or register.