FTD Fluid Markers for Degeneration: Check. For Pathology: Not Yet.
Quick Links
As the frontotemporal lobar degeneration field approaches an era of prevention and clinical trials, it needs robust biomarkers that herald the onset of symptoms and track the progression of a person’s underlying disease. Those tools are essential to developing effective treatments. Alas, the complexity of this group of rare diseases confounds the search, and international cohort studies such as Europe’s GENFI and North America’s ALLFTD are grappling with the challenge (see Part 1 of this series). Besides sitting for cognitive tests and questionnaires at every visit, participants with FTD and their asymptomatic family members are undergoing repeated lumbar punctures, blood draws, and brain scans for these longitudinal studies. Nearly a decade in, their efforts are starting to pay off in the form of antecedent markers that change preclinically and some that might hold promise for trials. Alas, with regard to disease-specific molecular markers, à la Aβ and phospho-tau for Alzheimer’s: No dice.
- Neurodegeneration markers NfL and NPTX2 change prior to symptoms in people with familial FTD.
- In initial clinical trials, CSF progranulin and poly-DP from C9ORF72 expansions changed in response to treatment. A lysosomal lipid looks promising, too.
- Urgently wanted: markers of TDP-43 and tau pathophysiology.
In recent papers, and at the International Conference for FTD, held virtually March 3–5, researchers shared some success stories and set their sights on still-elusive fluid markers—those that reveal the neuropathology lurking in the brain. They offered glimpses of theragnostic biomarkers—i.e., those that suggest a treatment may have met its target. In carriers of a C9ORF72 hexanucleotide expansion, poly-GP dipeptides plummeted in the CSF in response to a C9-targeted drug. For GRN mutation carriers, CSF progranulin rose in response to treatment, and a marker of lysosomal dysfunction tracked with symptom onset.
The best-validated fluid biomarker for FTD is a rather generic one: neurofilament light. NfL’s rap sheet dates back a quarter century, showing that this axonal protein trends upward in many neurodegenerative diseases (Rosengren et al., 1996; Alzbiomarker). GENFI and ALLFTD have established that NfL levels rise in both CSF and plasma prior to symptoms, and that plasma NfL reliably distinguishes between symptomatic and asymptomatic people (Sep 2016 news; van der Ende et al., 2019; Illán-Gala et al., 2021).
A newer marker of mayhem is neuronal pentraxin-2. NPTX2 is a synaptic protein whose concentration drops in CSF as synapses are lost in the brain. A recent GENFI paper reported NPTX2 inching downward in presymptomatic mutation carriers who subsequently became symptomatic (van der Ende et al., 2020). Glial fibrillary acidic protein—a marker of astrogliosis—also recently emerged as a promising CSF biomarker, at least among GRN mutation carriers. In them, GFAP was higher in symptomatic patients and tracked with brain atrophy and measures of symptom severity (Heller et al., 2020). John van Swieten of Erasmus University in Rotterdam led GENFI’s NfL and NPTX2 research; he believes pentraxin2 changes before NfL does, though larger studies still need to confirm this.
Van Swieten noted that these biomarker discoveries rest squarely on the shoulders of a small number of initially presymptomatic mutation carriers who develop symptoms while being tracked. So far in GENFI, that is the case for only nine participants. As a group, presymptomatic mutation carriers do not have significantly different levels of NfL, NPTX2, or other biomarkers compared to noncarriers, he said. The correlations will become clearer as the studies increase in size and length, and more people become symptomatic.
In the meantime, Bayesian statistics might help. At ICFTD, Adam Staffaroni of the University of California, San Francisco, reported results from a disease progression model. To span the spectrum, it incorporates biomarker measurements from mutation carriers across FTLD, rather than relying on individual longitudinal data. Staffaroni wove data from ALLFTD and GENFI, on 677 mutation carriers and 372 noncarriers, into his model to get a view of plasma NfL along with a handful of neuroimaging and cognitive markers. Plasma NfL appeared elevated years before symptoms emerged. This was most pronounced in GRN carriers, whose NfL concentrations were an order of magnitude above those of noncarriers a decade before clinical disease started, and ramped up dramatically afterward. Plasma NfL far outperformed neuroimaging or cognitive measures in terms of how dynamically it changed prior to symptoms.
This is not lost on drug developers. The biotech company Alector is putting plasma NfL to use as a screening tool in its Phase 2 and 3 trials of AL001, an anti-sortilin antibody meant to keep progranulin around longer in brain cells. In addition to symptomatic GRN mutation carriers, the company is selecting presymptomatic GRN mutation carriers with elevated NfL, on the assumption they are nearing onset and that a drug effect will be measurable in them. Whether NfL can also serve as a gauge of treatment responsiveness in FTD is unclear. That said, NfL did fall in successful treatment trials of spinal muscular atrophy and multiple sclerosis (Olsson et al., 2019; Sejbaek et al., 2019).
How about mutation-specific markers to track target engagement in clinical trials aimed at different forms of familial FTD? In carriers of C9ORF72 hexanucleotide expansions, poly-GP dipeptides that get translated off these repeats are indeed detectable in the CSF (Meeter et al., 2018). What’s more, the dipeptides plummet in response to C9-targeted treatment, according to data presented at ICFTD by Adam Boxer, University of California, San Francisco.
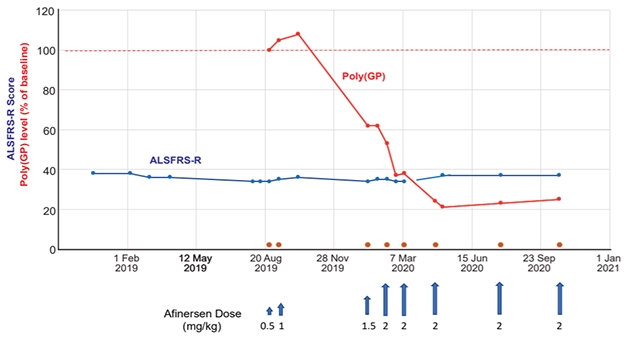
Dipeptide Pilot. In response to infusions (blue arrows) with a C9ORF72 -targeted antisense oligonucleotide therapy called afinersen, CSF poly-GP concentration plummeted, while symptoms remained stable, in a man with ALS. [Courtesy of Research Square preprint platform.]
At ICFTD, Boxer presented preliminary data, currently posted on the internet in preprint form, from Robert Brown’s group at the University of Massachusetts in Worcester (Tran et al., 2021). In a pilot experiment, the researchers treated a C9ORF72 expansion carrier who had atypical ALS with a C9ORF72-targeted anti-sense oligonucleotide. Called afinersen, this ASO is meant to silence expression of expanded C9ORF72 allele (Liu et al., 2021).
In response to intrathecal infusion of this ASO, poly-GP dropped in the man’s CSF by about 80 percent. This marker remained low, while the patient’s symptoms remained stable, over more than a year of eight infusions between August 2019 and October 2020. Curiously, the man’s CSF NfL levels rose, only dipping back down toward baseline levels once infusions were spaced further apart. The scientists suggested that this uptick in the neuronal injury marker might reflect nerve irritation provoked by the infusion.
In GRN mutation carriers, the CSF progranulin concentration serves as a biomarker. It is roughly half that in noncarriers, because these heterozygous mutations douse expression of the gene. Data from Alector’s trials suggests their anti-sortilin antibody restores progranulin levels to normal, and also reduces levels of plasma NfL (Aug 2020 conference news; Haynes et al., 2020).
In addition to progranulin itself, markers of lysososmal dysfunction resulting from progranulin deficiency might serve as disease markers, according to data presented by Isabelle Le Ber of Pitié-Salpêtrière University Hospital in Paris. Progranulin is processed into its active forms—granulin peptides—only within the lysosome. But progranulin does not travel to the lysosome unaccompanied; rather, it links up with prosaposin, another protein that relies on lysosomal cathepsins to unleash its active forms, saposins A, B, C, D.
At ICFTD, Le Ber explained that when progranulin is in short supply—as in GRN mutation carriers—fewer prosaposin proteins make it to the lysosome, leading to a build-up of the lysosphingolipids (lysoSPLs) that they process (Zhou et al., 2017). These excess lipids can be detected in the plasma of people with lysosomal storage disorders (Polo et al., 2019). At ICFTD, Le Ber reported that plasma lysoSPL was significantly elevated among 101 symptomatic GRN carriers compared to 27 presymptomatic carriers or 70 controls. The findings raise the possibility that the lysosomal marker could track with disease progression in GRN carriers.
Perhaps surprisingly, mutation-specific biomarkers have remained elusive for MAPT-FTD. In AD, several different plasma and CSF species of phospho-tau are wildly successful these days; alas, to date, no fluid or imaging tools detect the forms of tau that accumulate in FTLD. On the contrary, p-tau biomarkers that help diagnose AD are used to rule out AD in people with FTD. This is somewhat helpful given that people with FTD are commonly misdiagnosed as having AD (Mar 2020 news). Still, FTD researchers really would like their own tau markers to rule in FTD. At ICFTD, Henrik Zetterberg, University of Gothenburg, Sweden emphasized the need for such markers. So far, he confirmed, only the amyloid-dependent form of tau aggregation that happens in AD is detectable in body fluids.
A biomarker for FTLD-tau would be especially useful in cases of sporadic FTLD, roughly half of which have underlying tau pathology. Without a causal mutation, clinicians currently have no way to tell whether a given person with sporadic FTD has tau or TDP-43 pathology in their brain. Such neuropathological biomarkers are the Holy Grail for sporadic FTD, Boxer said. “Now we can rule out AD with a blood test. If we could identify who had underlying tau pathology and who had TDP-43, then we would be in great shape for clinical trials in sporadic FTD,” he said at ICFTD.
Zetterberg said that it will be challenging to develop biomarkers for TDP-43. In people with FTD, this protein ditches its home in the nucleus and accumulates in the cytoplasm. Recent studies have demonstrated that, similar to other proteopathic proteins such as tau and α-synuclein, TDP-43 can behave like a prion, propagating by way of templated misfolding (Jul 2010 news; Oct 2018 news). Taking advantage of this attribute, a recent study used real-time quaking-induced conversion (rtQuIC)—a method that detects prion-like seeds—to detect misfolded TDP-43 in the CSF of people with ALS or FTD who had TDP-43 pathology (Scialò et al., 2020; Orrú et al., 2011).
Similar tests are able to detect α-synuclein seeds in people with PD, tau in people with Pick’s disease, and, of course, prion proteins in people with prion disease (Oct 2020 news; Saijo et al., 2017; Dec 2016 news).
This type of assay requires a high level of expertise to do properly; still, Zetterberg was optimistic that something like it could pick up TDP-43 pathology. Boxer expressed interest, as well, though he noted that similar assays for tau would need to become more sensitive to be useful. Zetterberg and Boxer agreed that these markers might work to distinguish between people with tau versus TDP-43 pathology, yet might not be quantitative enough to track progression of pathology or response to treatment. —Jessica Shugart
References
News Citations
- Merged Consortia Forge Path to Trials in Frontotemporal Dementia
- Fluid NfL Shines, Tau PET Dims, in the Hunt for FTD Biomarkers
- At AAIC, Hints of Target Engagement for Some Drug Candidates
- A Phospho-Tau Plasma Assay for Alzheimer’s?
- Toxic TDP-43 Too Tough to Degrade, Plays Prion?
- TDP-43 Joins Cell-To-Cell Propagation Gang
- No Skin Off Your Nose: New Way to Diagnosing Parkinson’s?
- Methods to Detect Amyloid Seeds Improve, Extend to Blood and Parkinson’s
Biomarker Meta Analysis Citations
Paper Citations
- Rosengren LE, Karlsson JE, Karlsson JO, Persson LI, Wikkelsø C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem. 1996 Nov;67(5):2013-8. PubMed.
- van der Ende EL, Meeter LH, Poos JM, Panman JL, Jiskoot LC, Dopper EG, Papma JM, de Jong FJ, Verberk IM, Teunissen C, Rizopoulos D, Heller C, Convery RS, Moore KM, Bocchetta M, Neason M, Cash DM, Borroni B, Galimberti D, Sanchez-Valle R, Laforce R Jr, Moreno F, Synofzik M, Graff C, Masellis M, Carmela Tartaglia M, Rowe JB, Vandenberghe R, Finger E, Tagliavini F, de Mendonça A, Santana I, Butler C, Ducharme S, Gerhard A, Danek A, Levin J, Otto M, Frisoni GB, Cappa S, Pijnenburg YA, Rohrer JD, van Swieten JC, Genetic Frontotemporal dementia Initiative (GENFI). Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019 Dec;18(12):1103-1111. PubMed.
- Illán-Gala I, Lleo A, Karydas A, Staffaroni AM, Zetterberg H, Sivasankaran R, Grinberg LT, Spina S, Kramer JH, Ramos EM, Coppola G, La Joie R, Rabinovici GD, Perry DC, Gorno-Tempini ML, Seeley WW, Miller BL, Rosen HJ, Blennow K, Boxer AL, Rojas JC. Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease. Neurology. 2021 Feb 2;96(5):e671-e683. Epub 2020 Nov 16 PubMed.
- van der Ende EL, Xiao M, Xu D, Poos JM, Panman JL, Jiskoot LC, Meeter LH, Dopper EG, Papma JM, Heller C, Convery R, Moore K, Bocchetta M, Neason M, Peakman G, Cash DM, Teunissen CE, Graff C, Synofzik M, Moreno F, Finger E, Sánchez-Valle R, Vandenberghe R, Laforce R Jr, Masellis M, Tartaglia MC, Rowe JB, Butler CR, Ducharme S, Gerhard A, Danek A, Levin J, Pijnenburg YA, Otto M, Borroni B, Tagliavini F, de Mendonca A, Santana I, Galimberti D, Seelaar H, Rohrer JD, Worley PF, van Swieten JC, Genetic Frontotemporal Dementia Initiative (GENFI). Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020 Jun;91(6):612-621. Epub 2020 Apr 9 PubMed.
- Heller C, Foiani MS, Moore K, Convery R, Bocchetta M, Neason M, Cash DM, Thomas D, Greaves CV, Woollacott IO, Shafei R, Van Swieten JC, Moreno F, Sanchez-Valle R, Borroni B, Laforce R Jr, Masellis M, Tartaglia MC, Graff C, Galimberti D, Rowe JB, Finger E, Synofzik M, Vandenberghe R, de Mendonca A, Tagliavini F, Santana I, Ducharme S, Butler CR, Gerhard A, Levin J, Danek A, Frisoni G, Sorbi S, Otto M, Heslegrave AJ, Zetterberg H, Rohrer JD, GENFI. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020 Mar;91(3):263-270. Epub 2020 Jan 14 PubMed.
- Olsson B, Alberg L, Cullen NC, Michael E, Wahlgren L, Kroksmark AK, Rostasy K, Blennow K, Zetterberg H, Tulinius M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol. 2019 Sep;266(9):2129-2136. Epub 2019 May 23 PubMed.
- Sejbaek T, Nielsen HH, Penner N, Plavina T, Mendoza JP, Martin NA, Elkjaer ML, Ravnborg MH, Illes Z. Dimethyl fumarate decreases neurofilament light chain in CSF and blood of treatment naïve relapsing MS patients. J Neurol Neurosurg Psychiatry. 2019 Dec;90(12):1324-1330. Epub 2019 Oct 13 PubMed.
- Meeter LH, Gendron TF, Sias AC, Jiskoot LC, Russo SP, Donker Kaat L, Papma JM, Panman JL, van der Ende EL, Dopper EG, Franzen S, Graff C, Boxer AL, Rosen HJ, Sanchez-Valle R, Galimberti D, Pijnenburg YA, Benussi L, Ghidoni R, Borroni B, Laforce R Jr, Del Campo M, Teunissen CE, van Minkelen R, Rojas JC, Coppola G, Geschwind DH, Rademakers R, Karydas AM, Öijerstedt L, Scarpini E, Binetti G, Padovani A, Cash DM, Dick KM, Bocchetta M, Miller BL, Rohrer JD, Petrucelli L, van Swieten JC, Lee SE. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. 2018 May;5(5):583-597. Epub 2018 Apr 6 PubMed.
- Liu Y, Dodart JC, Tran H, Berkovitch S, Braun M, Byrne M, Durbin AF, Hu XS, Iwamoto N, Jang HG, Kandasamy P, Liu F, Longo K, Ruschel J, Shelke J, Yang H, Yin Y, Donner A, Zhong Z, Vargeese C, Brown RH Jr. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. 2021 Feb 8;12(1):847. PubMed.
- Haynes BA, Rhinn H, Yeh FL, Long H, Ward M, Hagey M, Siddiqui O, Paul R. AL001 restores CSF PGRN levels and normalizes disease‐associated biomarkers in individuals with frontotemporal dementia due to heterozygous mutations in the progranulin gene. Alzheimer's & Dementia, 7 December 2020
- Zhou X, Sun L, Bracko O, Choi JW, Jia Y, Nana AL, Brady OA, Hernandez JC, Nishimura N, Seeley WW, Hu F. Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat Commun. 2017 May 25;8:15277. PubMed.
- Polo G, Burlina AP, Ranieri E, Colucci F, Rubert L, Pascarella A, Duro G, Tummolo A, Padoan A, Plebani M, Burlina AB. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study. Clin Chem Lab Med. 2019 Nov 26;57(12):1863-1874. PubMed.
- Scialò C, Tran TH, Salzano G, Novi G, Caponnetto C, Chiò A, Calvo A, Canosa A, Moda F, Caroppo P, Silani V, Ticozzi N, Ratti A, Borroni B, Benussi L, Ghidoni R, Furlanis G, Manganotti P, Senigagliesi B, Parisse P, Brasselet R, Buratti E, Legname G. TDP-43 real-time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020;2(2):fcaa142. Epub 2020 Sep 14 PubMed.
- Orrú CD, Wilham JM, Raymond LD, Kuhn F, Schroeder B, Raeber AJ, Caughey B. Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. MBio. 2011;2(3):e00078-11. Print 2011 PubMed.
- Saijo E, Ghetti B, Zanusso G, Oblak A, Furman JL, Diamond MI, Kraus A, Caughey B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017 May;133(5):751-765. Epub 2017 Mar 14 PubMed.
Other Citations
Further Reading
Papers
- Holler CJ, Taylor G, Deng Q, Kukar T. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused byMutations. eNeuro. 2017 Jul-Aug;4(4) Epub 2017 Aug 18 PubMed.
- Illán-Gala I, Lleo A, Karydas A, Staffaroni AM, Zetterberg H, Sivasankaran R, Grinberg LT, Spina S, Kramer JH, Ramos EM, Coppola G, La Joie R, Rabinovici GD, Perry DC, Gorno-Tempini ML, Seeley WW, Miller BL, Rosen HJ, Blennow K, Boxer AL, Rojas JC. Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease. Neurology. 2021 Feb 2;96(5):e671-e683. Epub 2020 Nov 16 PubMed.
Primary Papers
- Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright C, Pinto C, Metterville J, Shin M, Sanil N, Dooley C, Puri A, Weiss A, Wightman N, Gray-Edwards H, Marosfoi M, King RM, Kenderdine T, Fabris D, Bowser R, Watts JK, Brown RH. Potent Mixed Backbone Antisense Oligonucleotide Safely Suppresses Expression of Mutant C9ORF72 Transcripts and Polydipeptides: First in Human Pilot Study. Research Square preprint, February 21 2021. Research Square.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.