CTE: Trauma Triggers Tauopathy Progression
Quick Links
What exactly goes on in the brains of people when blows to the head they received during sports or military service turn into a degenerative tau disease months or even years after the trauma? The answer to this question is unknown. At a conference on chronic traumatic encephalopathy (CTE) held this fall in Las Vegas, Nevada, a unique session combined human pathology with mouse model data (see Part 4 of this series) to showcase the cutting edge of what is being done to find out. First, Ann McKee of Boston University. “Ann’s research has been transformative. Without the pathologic basis she gave us, we would not know what to think about these cases,” said Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health, who hosted the conference. “Ann has examined more cases of CTE than any other pathologist in the world,” said Robert Cantu of Boston University.
In 2007, McKee started postmortem studies of concussed athletes and military veterans who donated their brains to the brain bank at BU’s Center for the Study of CTE, or whose families granted permission for autopsy. A review of the CTE pathology literature showed that most described cases up until recently were of boxers (McKee et al., 2009). By now, the majority of the BU samples are football players from high school to the NFL, as well as hockey players and military veterans. Notably, athletes without CTE symptoms have begun pledging their brains to the brain bank in order to provide controls and put the collection on a broader footing.
To date, 136 brains have come into the center’s brain bank, and approximately 100 have been analyzed thus far. About 80 percent had CTE. In Las Vegas, McKee updated the audience on her results. The postmortem pathology of CTE is unique. The gross appearance of the brain changes as the disease advances. Ventricles 2 and 3 expand, and the medial temporal lobe shrinks, as seen in Alzheimer’s. Unlike in AD, the septum pellucidum—a single membrane separating the left and right lateral ventricles—is perforated. Most striking about CTE is the extent and distinctive nature of its tauopathy, McKee said. Hyperphosphorylated tau forms deposits in an irregular, patchy distribution. Curiously, it is densest at the bottom curves of the cortical ribbon, where it engulfs blood vessels. “We see tremendous abnormality at the depth of the sulci,” McKee said, “The sulci are the epicenters of this pathology.”
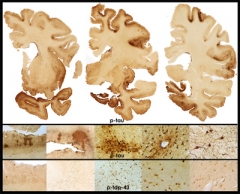
Stage 4 CTE
in a 65-year-old former professional football player is evidenced by extensive atrophy and irregular deposits of phospho-tau and TDP-43 proteins throughout frontal and temporal cortices and deep nuclei. Image courtesy of Ann McKee, Boston University School of Medicine/VA Boston.
McKee insists that CTE pathology is distinct from AD. “Most CTE cases I see have no β amyloid, and if they do, then a low amount of diffuse plaques,” McKee said. Earlier studies have reported β amyloid pathology following head trauma (e.g., Roberts et al., 1990; Graham et al., 1995; Roberts et al., 1994). These studies looked early—in some cases only hours or days—in people who had suffered severe injuries, not years after repeated mild injuries. This older work interprets the amyloid deposition as resulting from an acute-phase APP upregulation in response to the trauma. CSF studies have been consistent with that (Olsson et al., 2004). This literature supported the established conclusion that head injury is a risk factor for dementia (for definitive discussion, see AlzRisk meta-analysis).
In McKee’s series, CTE looks like a progressive tauopathy that expands with age. Starting out in frontal cortex, it spreads to insular and temporal cortex, then amygdala and hippocampus. As do frontotemporal dementias and Parkinson’s, CTE also affects subcortical structures such as the thalamus, the hypothalamus, and brainstem, the locus ceruleus and substantia nigra. “There is marked tau deposition throughout the brain,” McKee said. In addition, CTE frequently features TDP-43 deposition, again in a unique distribution, and axonal damage.
McKee is developing a staging system for CTE. As an example for Stage 1, she showed brain slices of a high school football player who died at age 18. “I was very surprised to find tau foci in his frontal cortex,” McKee said. She also showed tau foci in the brain of a 21-year-old team captain in college who hanged himself in his apartment a day after telling his parents he worried about his academics. Stage 1 was apparent in the brain of a U.S. Marine veteran who had shot himself at 28. He had had multiple concussions from a bicycle accident, football practice, combat deployments, and a motor vehicle accident, McKee said. He had had anxiety, as well as trouble concentrating and finding words. Stage 2 features multiple perivascular tauopathy foci and greater evidence of axonal loss. As examples, McKee cited the case of NHL player Derek Boogaard, an enforcer who served 589 penalty minutes in his career. Nicknamed Boogeyman for his toughness, he struggled with depression, and memory and concentration problems, though stays in drug rehab made it difficult to relate causes and symptoms. Boogaard died at 28 (see NYT video series). Similar CTE pathology occurred in the brain of a 45-year-old Operation Enduring Freedom/Operation Iraqi Freedom army veteran who had been exposed to a single close-range IED blast, McKee told the audience in Las Vegas.
As an example of Stage 3, which is marked by further spread of tau deposits and axonopathy, McKee showed the case of Dave Duerson, who shot himself in the chest after leaving a note requesting that his family donate his brain to research. Stage 4 represents endstage CTE. McKee showed the brain of an 80-year-old Football Hall of Famer who was also an army veteran. His septum was broken and the brain shrunken, the medial-temporal lobe a third of its normal age-controlled weight. “Even he had no β amyloid, but extreme tauopathy everywhere in the brain,” McKee said. At each stage, some athletes and blast-exposed veterans appeared indistinguishable pathologically.
Like most neurodegenerative diseases, CTE comes in variants. Of the current set of 68 brains, 51 represent pure CTE, but eight were a combination of CTE and motor neuron disease. The other nine included multiple pathologies that met diagnostic criteria for AD, Parkinson’s/dementia with Lewy bodies (DLB), or frontotemporal dementia (FTD). The ALS-like disorder has drawn particular attention (see ARF related news story). McKee cited the case of a college football player who had started the sport at age eight and had sustained many concussions. He contacted McKee and Chris Nowinski at Boston University, who co-founded the Center for the Study of CTE, and shortly before he died from ALS-like symptoms, asked that his brain be studied. It showed profound CTE and TDP-43 proteinopathy in the spinal cord.
This pathology suggests that CTE and its variants may underlie some clinical diagnoses of AD, PD, and ALS given to former athletes and soldiers who display symptoms of these better-known diseases, said Cantu. At the Lou Ruvo conference, Cantu cited a widely noted study that reported initial epidemiological numbers for the incidence of Alzheimer’s, Parkinson’s, and ALS in former NFL players (see ARF related news story). This study represents the best incidence numbers so far, but even so, it may be skewed in that some of those diagnoses, made without autopsy confirmation, probably represent CTE and its variants, Cantu said. The study analyzed death certificates, not the brains of the players. As such, it is also subject to undercounting, as causes of death are often incompletely noted on these forms. “I have filled out many death certificates over the years. In the distress of the situation, doctors are often less detailed than they could be,” Cantu said, “We still do not know the incidence and prevalence of CTE. Death certificates are a start, but we need to study brains and use autopsy-confirmed diagnoses.”—Gabrielle Strobel.
This is Part 3 of a six-part series. See also Part 1, Part 2, Part 4, Part 5, Part 6. Read a PDF of the entire series.
References
News Citations
- Do Tau "Prions" Lead the Way From Concussions to Progression?
- TDP-43 and Tau Entangle Athletes’ Nerves in Rare Motor Neuron Disease
- Dementia Four Times More Likely in Pro Football Players
- Meet the New Progressive Tauopathy: CTE in Athletes, Soldiers
- Boxing: Study of Human Model for CTE Enters Second Round
- CTE Needs Consensus on Lifetime Diagnosis
- CTE Advocates Pivot Toward Preventing Concussions in Kids
Paper Citations
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009 Jul;68(7):709-35. PubMed.
- Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990 May;53(5):373-8. PubMed.
- Graham DI, Gentleman SM, Lynch A, Roberts GW. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995 Feb;21(1):27-34. PubMed.
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994 Apr;57(4):419-25. PubMed.
- Olsson A, Csajbok L, Ost M, Höglund K, Nylén K, Rosengren L, Nellgård B, Blennow K. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol. 2004 Jul;251(7):870-6. PubMed.
Other Citations
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.