In Familial Alzheimer’s, Tau Creeps into Cortex as Symptoms Show
Quick Links
Researchers led by Yakeel Quiroz at Massachusetts General Hospital in Boston have used PET imaging to measure both Aβ and tau deposition in members of a large Colombian kindred affected by an autosomal-dominant Alzheimer’s disease mutation. On average, family members who inherit the mutation develop symptoms by the age of 44. In the February 12 JAMA Neurology, Quiroz and colleagues report that Aβ starts to accumulate in the neocortices of carriers 10 to 15 years prior, whereas tau deposits emerge in the medial temporal lobe about six years prior and spread from there into the cortex even later, once people start to have trouble with memory. The new findings support the idea that Aβ instigates the spread of tau pathology beyond the medial temporal lobe, and that this spreading process is intimately connected with the cognitive manifestations of AD. Co-author Keith Johnson and colleagues recently came to the same conclusion after analyzing longitudinal data from people with late-onset AD (Feb 2018 news).
- A cross-sectional study measured Aβ, tau, and cognition in the Colombian PSEN1 E280A kindred.
- In carriers, Aβ rose 15 years prior to expected symptom onset, while tau rose up to six years prior in the medial temporal lobe.
- Tau pathology invaded the cortex mostly in symptomatic people.
“This [study] helps us to better understand the preclinical trajectory of this early onset autosomal-dominant disease tightly linked to amyloid overproduction as its driving force,” commented Samuel Lockhart of Wake Forest University in Winston-Salem, North Carolina.
“This study adds to a growing appreciation for the utility of biomarkers in identifying preclinical AD,” said Samantha Burnham of Commonwealth Scientific and Industrial Research Organization in Canberra, Australia. She added that the close association between tau accumulation and cognitive symptoms suggests that tau PET imaging might be a useful marker of treatment response in clinical studies.
Research from several groups has shown conclusively that the pathological process of AD begins many years prior to the first signs of forgetfulness. Understanding more about the preclinical course of the disease is crucial for the development of biomarkers and effective treatments. People with autosomal-dominant AD mutations are destined to have AD, and people who inherit the same mutation tend to have similar ages at onset. An estimated 1,800 people in and around the Colombian city of Medellín carry the PSEN1 E280A mutation. Most develop mild cognitive impairment (MCI) due to AD between the ages of 43–45 and dementia before they turn 50 (Acosta-Baena et al., 2011). Hundreds of these family members have volunteered for longitudinal studies, which have yielded invaluable information on AD biomarkers (Jan 2015 news).
In the current study, Quiroz added tau PET imaging to the roster of analyses, making this the largest tau-PET imaging study on people with a single ADAD mutation to date. Twenty-four members of the kindred traveled from Colombia to Boston to undergo brain scans at Massachusetts General Hospital. They averaged 38 years old, and included 12 cognitively normal noncarriers, as well as 12 carriers, of whom nine were cognitively normal and three had MCI.
The researchers first assessed levels of Aβ accumulation based on the distribution volume ratio (DVR) of the PET ligand PiB, using the cerebellum as a reference. None of the noncarriers had any signs of elevated Aβ, nor did the youngest carrier, who was 28. The other 11 carriers all had evidence of Aβ accumulation, with seven reaching the brain-wide threshold for positivity—a PiB DVR of at least 1.2. The extent of Aβ accumulation increased with advancing age of the carriers, and the three carriers with MCI had the highest burden of Aβ. The plaque distribution resembled patterns reported in people with late-onset AD (LOAD), with preferential PiB binding in the posterior cingulate, precuneus, parietotemporal, frontal, and basal ganglia regions.
Quiroz next assessed tau accumulation, comparing the average standard uptake value ratios (SUVR) of 18F-flortaucipir, formerly called AV1451, in different regions of the brain. She told Alzforum that while the field has not yet agreed upon a standard threshold for tau accumulation, they chose an SUVR of 1.3 as a working threshold for this study. None of the noncarriers showed signs of elevated tau deposition in the medial temporal lobe (mTL) or elsewhere, regardless of their age. Tau deposits are often detected in the mTL of aging people; however, all of the noncarriers were 55 or younger, most in their 30s or 40s.
Likewise, little flortaucipir stuck in the mTL of the six mutation carriers who were under age 38. However, five of the six carriers who were 38 or older had evidence of tau accumulation in the mTL. Of these five, four also had substantial tau deposition in the cortex, most notably in the inferior and lateral temporo-parietal, the parieto-occipital, the posterior cingulate cortex, and in the precuneus. The three people with MCI had cortical tau, as did one cognitively normal mutation carrier. People with elevated cortical tau also had the highest degree of cortical Aβ accumulation.
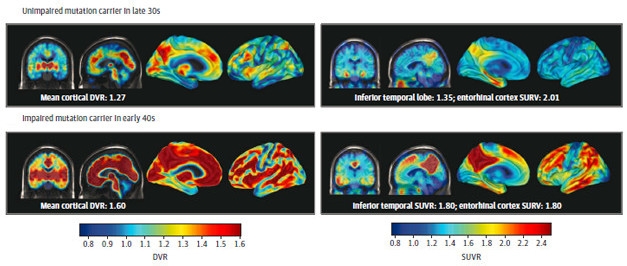
Aβ Begets Tau. PET imaging of Aβ (left panels) and tau (right panels) in unimpaired (top) and impaired (bottom) mutation carriers. Both pathologies become more extensive after symptoms appear. [Courtesy of Quiroz et al., JAMA Neurology, 2018.]
Among mutation carriers, the amount of tau accumulation in the entorhinal cortex and in the inferior temporal lobe correlated with poorer performance on memory tests, and this held true even among the nine presymptomatic carriers.
“By studying mutation carriers at various stages of the disease cascade, the authors provide one of the first comprehensive views of the pattern of AV1451 binding in ADAD,” commented Eric McDade of Washington University in St. Louis. “Importantly, this work supports many of the studies in sporadic AD by indicating that AV1451 binding is absent in those without fibrillar Aβ plaques, and that once symptoms begin, AV1451 binding increases in limbic/paralimbic cortices and then spreads to heteromodal cortices with increased symptoms.”
While Quiroz’s analysis is the largest tau PET study on carriers of a single ADAD mutation, preliminary findings from the Dominantly Inherited Alzheimer’s Network (DIAN) reported tau distribution in 14 symptomatic carriers, 20 asymptomatic carriers, and 16 noncarriers (Aug 2017 conference news). Similar to Quiroz, the DIAN researchers also found that Aβ started accumulating more than a decade prior to estimated symptom onset, but the two studies differed slightly with respect to the timing of tau pathology in the mTL. In DIAN, that first began around the age at onset.
Why the difference? Quiroz speculated that by studying carriers of just a single AD mutation, she teased out the relationship between tau accumulation and disease stage more precisely than can be done in a more heterogeneous cohort comprising dozens of mutations in APP and the presenilins. Others noted that that small number of participants must be kept in mind. In Quiroz’s paper, three of the five mutation carriers with entorhinal tau accumulation were symptomatic, and one of the two asymptomatic carriers was younger than 40, leaving only one asymptomatic, tau-positive person who was six years away from age of onset. However, Quiroz told Alzforum that up to now, a total of 42 members of the Colombian kindred have had flortaucipir imaging to date, and the timing of tau accumulation in the mTL has held at around six years before disease onset.
To Michel Grothe of the German Center for Neurodegenerative Diseases in Rostock, the appearance of tau pathology in the mTL in people without neocortical tangles was the most striking result of the paper. He pointed out that the existence of age-related tauopathy in the mTL has blurred the connections between Aβ and mTL tau pathology in people with LOAD. However, in PSEN1 E280A carriers, any tau pathology can be assumed to be a direct consequence of the overproduction of Aβ caused by the mutation, since they are so young. “This underlines the high potential of cerebral amyloidosis to facilitate the accumulation of medial temporal tau pathology, even in the absence of initial age-related tau changes to act upon,” he wrote to Alzforum. “Given the strikingly low levels of amyloid deposits in the medial temporal lobe in both autosomal-dominant and sporadic forms of AD, this effect is likely to be mediated by remote mechanisms that remain to be elucidated.”
How does this PET data tie in with changes in soluble tau in the CSF? In a joint commentary alongside Quiroz’s paper, McDade and Randall Bateman, also at WashU, pointed out that in the DIAN cohort, the concentration of tau in the cerebrospinal fluid rises more than a decade before estimated symptom onset (Jul 2012 news). This early elevation of CSF tau has also been reported in the PSEN1 E280A carriers (Fleisher et al., 2015). “Taken together, these [CSF and PET] findings suggest a modified pathobiological cascade of AD, with tau in cerebrospinal fluid changing 10–15 years before neurofibrillary tau pathology begins,” they wrote.
Both the DIAN and Colombian tau PET imaging studies came to similar conclusions about cortical tau—it largely appeared in people with symptoms, and the precuneus and posterior cingulate took on heavy tau burdens, as did the inferior temporal region. A recent DIAN study also found that the precuneus was the earliest region to accumulate Aβ and to have metabolic and structural changes (Gordon et al., 2018). McDade therefore suggested that in the future, it will be important to understand how tau accumulation in these posterior regions relates to Aβ accumulation, and to specific changes in cognition. Quiroz added that longitudinal studies of the Colombian kindred will track the trajectory of tau in the cortex.
Will these findings in ADAD help researchers understand the trajectory of LOAD? Michael Donohue of the University of Southern California in San Diego believes that despite the obvious limitation of small sample size, Quiroz’s study supports a strong congruence between the two forms of AD. “The findings are strikingly consistent with prospective and retrospective studies of LOAD and other studies of ADAD,” he wrote. “Namely, the finding that Aβ pathology is apparent approximately 15 years prior to symptom onset, and tau pathologies are apparent approximately six years prior to symptom onset, is consistent with other studies in LOAD. The importance of this consistency is to support our hope that future findings in ADAD might translate to LOAD and vice versa.”
Lockhart thinks this study of young ADAD mutation carriers will help researchers distinguish between the effects of central AD pathology and the myriad comorbidities that affect people with LOAD. “Results like those presented here help demonstrate the background of amyloid-driven, tau-mediated neurodegeneration that may be present in patients with LOAD but may interact with other injurious processes to negatively impact cognition and function,” he wrote to Alzforum. “This enables us to be able to better investigate, for example, how elevated vascular risk factors may contribute to tau pathology in late life above and beyond the contributions of amyloid pathology.”—Jessica Shugart
References
News Citations
- Imaging Clinches Causal Connections between Aβ, Tau, Circuitry, and Cognition
- API Biomarker Data Mirror DIAN’s, Support Progression Models
- Data from DIAN Revise Familiar Biomarker Trajectories
- Paper Alert: DIAN Biomarker Data Show Changes Decades Before AD
Mutations Citations
Paper Citations
- Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, Saldarriaga A, Lopera F. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011 Mar;10(3):213-20. PubMed.
- Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gutierrez Gomez M, Langois CM, Langbaum JB, Roontiva A, Thiyyagura P, Lee W, Ayutyanont N, Lopez L, Moreno S, Muñoz C, Tirado V, Acosta-Baena N, Fagan AM, Giraldo M, Garcia G, Huentelman MJ, Tariot PN, Lopera F, Reiman EM. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 2015 Mar;72(3):316-24. PubMed.
- Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, Christensen J, McDade E, Wang G, Xiong C, Cairns NJ, Hassenstab J, Marcus DS, Fagan AM, Jack CR Jr, Hornbeck RC, Paumier KL, Ances BM, Berman SB, Brickman AM, Cash DM, Chhatwal JP, Correia S, Förster S, Fox NC, Graff-Radford NR, la Fougère C, Levin J, Masters CL, Rossor MN, Salloway S, Saykin AJ, Schofield PR, Thompson PM, Weiner MM, Holtzman DM, Raichle ME, Morris JC, Bateman RJ, Benzinger TL. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018 Mar;17(3):241-250. Epub 2018 Feb 1 PubMed.
Further Reading
Primary Papers
- Quiroz YT, Sperling RA, Norton DJ, Baena A, Arboleda-Velasquez JF, Cosio D, Schultz A, Lapoint M, Guzman-Velez E, Miller JB, Kim LA, Chen K, Tariot PN, Lopera F, Reiman EM, Johnson KA. Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol. 2018 May 1;75(5):548-556. PubMed.
- McDade E, Bateman RJ. Tau Positron Emission Tomography in Autosomal Dominant Alzheimer Disease: Small Windows, Big Picture. JAMA Neurol. 2018 May 1;75(5):536-538. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Commonwealth Scientific and Industrial Research Organisation
This is a unique study examining ADAD individuals with a single-gene (PSEN1) mutation. The well-characterized age of onset for mild cognitive impairment (MCI) and Alzheimer’s disease in the participants allowed for a crucial temporal assessment of Aβ and tau. Findings suggested elevated levels of neocortical Aβ occur over a decade prior to clinical symptoms (similar to reports in late-onset AD) and tau deposition occurs approximately six years prior to MCI onset. This study adds to a growing appreciation for the utility of biomarkers in identifying preclinical AD. The strong association between tau pathology and cognitive measures is of particular interest: Tau may add staging information indicative of the magnitude of change on clinical endpoints.
Wake Forest School of Medicine
There are two big reasons I like this paper and think it will shape research to come, not only in ADAD but also in LOAD.
First, it was very interesting to see how multimodal amyloid and tau PET biomarkers were associated with each other, with cognitive testing data, and with age/estimated year of onset in the Colombian (PSEN1 E280A) AD kindred. Quiroz and colleagues have nicely shown, using in vivo imaging, that in PSEN1 mutation carriers, amyloid pathology likely precedes tau pathology in the neocortex, and that after tau appears in the cortex that both measures continue to increase with respect to each other and to reduced cognitive performance. This helps us to better understand the preclinical trajectory of this early onset, autosomal-dominant disease, which is tightly linked to amyloid overproduction as its driving force.
However, what I find most interesting about this article is how it sets the stage for understanding how the older adult brain is besieged by multiple co-occurring disease processes—not just amyloid pathology—in late life. With advancing age, people present more often with not just amyloid and tau pathology but also vascular disease and other proteinopathies. While there are differences between ADAD and LOAD, results like those presented here (e.g., Figs. 1 and 2) help demonstrate the background of amyloid-driven, tau-mediated neurodegeneration that may be present in patients with LOAD but may interact with other injurious processes to negatively impact cognition and function. This enables us to better investigate, for example, how elevated vascular risk factors may contribute to tau pathology in late life above and beyond the contributions of amyloid pathology.
What I would be excited to see in terms of next steps are (1) a simultaneous examination of these ADAD carriers against LOAD patients with similar disease stage (e.g. CDR = 0.5) to directly compare PET patterns and other biomarkers, and (2) the associations between amyloid, tau, cognition, and MRI measures of neurodegeneration in this cohort.
University of California, San Diego
The authors are right to caution readers about the limitation of extrapolating this autosomal-dominant AD (ADAD) data to late-onset AD (LOAD). It is also a relatively small study of just 12 Presenilin 1 E280A mutation carriers and 12 noncarriers. Nevertheless, the findings are strikingly consistent with prospective and retrospective studies of LOAD and other studies of ADAD. Namely, the finding that Aβ pathology is apparent approximately 15 years prior to symptom onset, and tau pathologies are apparent approximately six years prior to symptom onset, is consistent with other studies in LOAD. The importance of this consistency is to support our hope that future findings in ADAD might translate to LOAD and vice versa.
Clearly there is more variability in the age of onset in LOAD than in ADAD, but some variability still exists in ADAD. This suggests that the correlation between years to symptom onset and pathology would be even stronger than the correlation between age and pathology. We might see this borne out in future studies with longitudinal data in this cohort and others. And ongoing efforts to improve the estimation of years to symptom onset, e.g. “latent disease time,” might facilitate more useful comparisons of LOAD and ADAD (Li et al., 2017).
References:
Li D, Iddi S, Thompson WK, Donohue MC, Alzheimer’s Disease Neuroimaging Initiative. Bayesian latent time joint mixed effect models for multicohort longitudinal data. Stat Methods Med Res. 2017 Jan 1;:962280217737566. PubMed.
German Center for Neurodegenerative Diseases (DZNE)
To me, one of the most interesting results of this exciting study is the clear initial appearance of tau in the medial temporal lobe without notable neocortical tau. Although the medial temporal lobe has long been recognized as the initial site of tau pathology in sporadic AD (Braak and Braak, 1991), this does not necessarily have to translate to genetically determined forms of the disease. While there is accumulating evidence that genetically determined and sporadic forms of AD follow similar pathogenetic trajectories when observed on global biomarker levels (i.e., amyloid then tau then neurodegeneration), there may be important regional differences in the onset and evolution of the different types of pathology. For example, studies analyzing amyloid-PET data on a regional level have reported a very early striatal amyloid deposition in autosomal-dominant AD that appears before the occurrence of neocortical amyloid (Klunk et al., 2007). This is in striking contrast to the late-stage striatal involvement in established amyloid-staging schemes for sporadic AD (Thal et al., 2002), which we could recently reproduce using a PET-based in-vivo staging approach (Grothe et al., 2017).
Moreover, recent data from the DIAN study indicates that neurodegeneration as measured by structural MRI, which tends to follow the regional pattern of tau accumulation with some temporal delay, may manifest first in the precuneus in this cohort (Gordon et al., 2018). Yet the present data by Quiroz et al. suggest that the analyzed PSEN1 mutation carriers show a very similar regional spread of tau pathology, from an initially affected medial temporal lobe to inferior temporal and posterior parietal neocortical areas, as would be expected from sporadic AD. The observation of frequent medial temporal tau deposits in elderly individuals without any evidence of amyloid pathology, a phenomenon termed primary age-related tauopathy, has raised questions as to the general relation between medial temporal tau accumulation and AD pathology (Jellinger et al., 2015; Duyckaerts et al., 2015).
In this context it has been proposed that it is the additional amyloid pathology in AD that drives tau out of the medial temporal lobe and into neocortical areas. The PSEN1 mutation that characterizes the analyzed cohort of the present study is causally linked to an aberrant cellular metabolism of Aβ, and thus any tau deposits observed in these mutation carriers at this young age can be reasonably assumed to be a direct downstream consequence of the primary amyloid pathology. This underlines the high potential of cerebral amyloidosis to facilitate the accumulation of medial temporal tau pathology, even in the absence of initial age-related tau changes to act upon. Given the strikingly low levels of amyloid deposits in the medial temporal lobe in both autosomal-dominant and sporadic forms of AD, this effect is likely to be mediated by remote mechanisms that remain to be elucidated.
References:
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, Dekosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007 Jun 6;27(23):6174-84. PubMed.
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002 Jun 25;58(12):1791-800. PubMed.
Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ, Alzheimer's Disease Neuroimaging Initiative. In vivo staging of regional amyloid deposition. Neurology. 2017 Nov 14;89(20):2031-2038. Epub 2017 Oct 18 PubMed.
Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, Christensen J, McDade E, Wang G, Xiong C, Cairns NJ, Hassenstab J, Marcus DS, Fagan AM, Jack CR Jr, Hornbeck RC, Paumier KL, Ances BM, Berman SB, Brickman AM, Cash DM, Chhatwal JP, Correia S, Förster S, Fox NC, Graff-Radford NR, la Fougère C, Levin J, Masters CL, Rossor MN, Salloway S, Saykin AJ, Schofield PR, Thompson PM, Weiner MM, Holtzman DM, Raichle ME, Morris JC, Bateman RJ, Benzinger TL. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018 Mar;17(3):241-250. Epub 2018 Feb 1 PubMed.
Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, Dickson DW, Hof PR, Hyman BT, Jack CR Jr, Jicha GA, Knopman DS, Kovacs GG, Mackenzie IR, Masliah E, Montine TJ, Nelson PT, Schmitt F, Schneider JA, Serrano-Pozo A, Thal DR, Toledo JB, Trojanowski JQ, Troncoso JC, Vonsattel JP, Wisniewski T. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015 May;129(5):757-62. Epub 2015 Mar 17 PubMed.
Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, Dickson DW, Hof PR, Hyman BT, Jack CR Jr, Jicha GA, Knopman DS, Kovacs GG, Mackenzie IR, Masliah E, Montine TJ, Nelson PT, Schmitt F, Schneider JA, Serrano-Pozo A, Thal DR, Toledo JB, Trojanowski JQ, Troncoso JC, Vonsattel JP, Wisniewski T. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015 May;129(5):757-62. Epub 2015 Mar 17 PubMed.
Washington University
Quiroz and colleagues provide the first report of AV1451 tau-PET in a cohort of at risk for autosomal-dominant Alzheimer's disease (ADAD) related to the PSEN1 E280A mutation. By studying mutation carriers at various stages of the disease cascade (amyloid-negative, amyloid-positive/asymptomatic, symptomatic) they provide one of the first comprehensive views of the pattern of AV1451 binding in ADAD. Importantly, this work supports many of the studies in sporadic AD by indicating that AV1451 binding is absent in those without fibrillar Aβ plaques, and that once symptoms begin, AV1451 binding increases in limbic/paralimbic cortices and then spreads to heteromodal cortices with increased symptoms. Besides being able to track AV1451 pathology across the AD spectrum with a small number of participants (24), this work clearly highlights the complexity of tau-related pathology and biology in AD. Specifically, previous work in this same population has suggested that CSF tau and p-tau increase up to 15 years before anticipated symptoms (Fleisher et al., 2015), but AV1451 only six years before. This work highlights the importance of considering the different measures of tau pathology as related, but distinct.
One limitation of this study was the restriction of regional AV1451 (F18 FTP) to the inferior temporal regions when examining the association with cortical PiB-PET and neuropsychological measures. The voxel-wise comparison of AV1451 between mutation carriers and noncarriers (as well as symptomatic and asymptomatic mutation carriers) indicated important differences in AV1451 binding in areas outside of the inferior temporal lobes as well. Therefore, it is possible additional findings between regional AV1451 and cognition or fibrillar amyloid may have been identified, or had stronger correlations with regions like the posterior-cingulate and precuneus. This is particularly important to consider, as preliminary data from the DIAN study indicates the precuneus as a region with early and robust AV1451 binding (Benzinger et al., 2016). With increased numbers of participants from this cohort and in the DIAN study, the differences and similarities between ADAD and sporadic AD as it relates to neurofibrillary tau pathology will become clearer.
References:
Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gutierrez Gomez M, Langois CM, Langbaum JB, Roontiva A, Thiyyagura P, Lee W, Ayutyanont N, Lopez L, Moreno S, Muñoz C, Tirado V, Acosta-Baena N, Fagan AM, Giraldo M, Garcia G, Huentelman MJ, Tariot PN, Lopera F, Reiman EM. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 2015 Mar;72(3):316-24. PubMed.
Make a Comment
To make a comment you must login or register.