Will RNA molecules that bump up or tamp down gene expression live up to their potential? Results from RNA-targeting therapies presented at the 70th annual meeting of the American Academy of Neurology in Los Angeles suggest children with spinocerebellar ataxia continue to improve years after starting approved treatments. However, prices are high and so far, health gains are modest. Will similar approaches for Huntington’s disease, ALS, familial amyloid polyneuropathy, and tauopathies fare better?
As RNA Therapies Come of Age, Efficacy Remains Weak
Recent approvals of antisense therapies for spinal muscular atrophy and Duchenne muscular dystrophy have bolstered hopes for targeting RNA to treat a plethora of neurodegenerative brain disorders. At the 70th annual meeting of the American Academy of Neurology, April 21 to 27 in Los Angeles, scientists heard the latest results about nusinersen, an FDA-approved antisense oligonucleotide (ASO) for the treatment of childhood spinal muscular atrophy, and about inotersen and patisiran, antisense- and siRNA-based drugs, respectively, for the treatment of transthyretin amyloidosis.
The data suggest patients can continue to improve years after starting treatment, and that the earlier they start, the better. Still, the treatments fall far short of cures and costs are staggering. Antisense treatments for Huntington’s, ALS, and tauopathies are in early stage trials and seem safe (see Part 2 of this story).
Steady but Small Improvement from Nusinersen Diana Castro, UT Southwestern Medical Center, Dallas, reviewed new clinical data on nusinersen. Ionis Pharmaceuticals of Carlsbad, California, designed this ASO to compensate for mutations in the gene for survival motor neuron 1 (SMN1). By altering gene splicing, nusinersen bolsters production of the full-length version of the nearly identical SMN2. The ASO made headlines in 2016 when interim analyses of two Phase 3 trials revealed that children taking the drug had improved on tests of motor function and had better odds of surviving (Finkel et al., 2017; Mercuri et al., 2018). The trials were stopped to allow all children to switch to the drug in an open-label extension trial (Nov 2016 news).
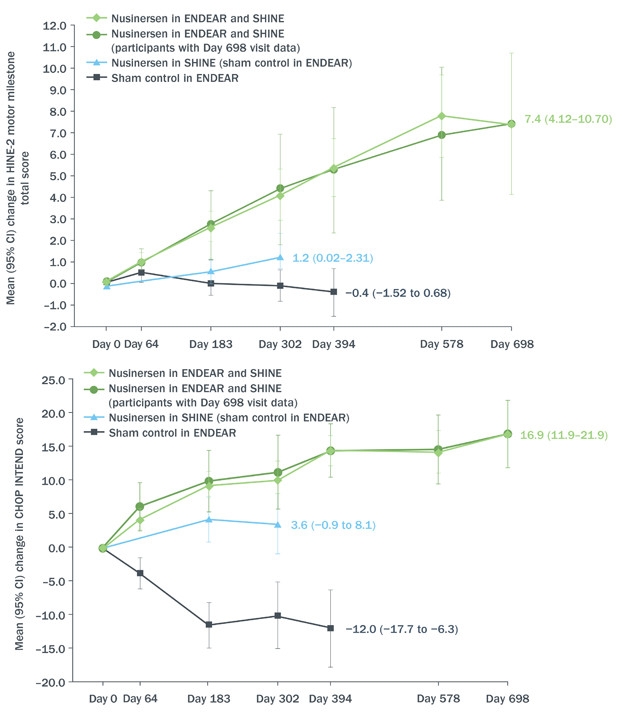
Castro reviewed data from the Phase 3 ENDEAR study combined with interim results as of June 30, 2017, from the open-label extension SHINE (see image below). ENDEAR enrolled 122 babies who had been diagnosed as most likely having SMA type 1, the severest kind. Of the 81 who received nusinersen, 65 transitioned into SHINE, as did 24 of 41 who had been on placebo. All started their intervention when they were seven months old or younger. In both ENDEAR and SHINE, nusinersen was delivered into the cerebrospinal fluid. Four loading injections a few weeks apart were followed by maintenance injections every four months. Each injection delivered 12 mg of nusinersen.
Still Going. From day zero of open-label extension, babies who received nusinersen at seven months or younger (green) continued to benefit according to HINE-2 (top) and CHOP-INTEND (bottom) neurological tests of infant development. Those who switched from placebo to drug around 18 months of age improved, as well (blue), at least compared to their prior deterioration during the placebo-controlled phase of the trial (black). [Courtesy Diana Castro, UT Southwestern Medical Center.]
Of the 81 babies who started treatment in ENDEAR, 12 could sit independently, and 23 were able to control their head movements fully at the time of their last assessment—for some, nearly three years after receiving their first dose. Sitting and head control are developmental milestones that babies with SMA1 are not expected to achieve. Although none could stand or walk independently, mean motor function had continued to improve for the group, changing by 5.8 on Section 2 of the Hammersmith Infant Neurological Examination. This measure of infant motor development assigns scores of zero to four in eight categories, from controlling head movement to sitting, kicking, crawling, and eventually walking. Clinicians deemed 51 of the 81 infants “responders,” based on their scores in individual categories of the HINE-2. Responders were those who improved in more categories than they worsened, or improved by one or more points in six of the eight categories on the scale. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND), a similar test designed for evaluating very weak kids, indicated improvement in motor function in 55 patients.
The drug extended life. The median time to death or respiratory failure for infants who received nusinersen at seven months or younger was 73 weeks, compared with 23 weeks for those who started in the placebo group.
Moreover, although 12 of the 24 babies who had received placebo in ENDEAR needed permanent respirator assistance by the start of the extension, this group as a whole seemed to stabilize during SHINE, registering a small mean improvement of 1.1 on the HINE-2, and four children qualified as responders. However, of the 12 children who started SHINE free of permanent respirator assistance, five had died or gone on permanent ventilation.
In discussion, John Day, Stanford University, and Richard Finkel, Nemours Children’s Hospital in Orlando, Florida, noted that other studies of nusinersen point to the potential for improvement in older children or even in adults with SMA. “There may be a small pool of more resilient motor neurons that can be rescued,” suggested Finkel.
Consistent with results from previous trials, there were no serious events in SHINE that seemed related to treatment. Fever and upper respiratory tract infections were the most frequent adverse events.
Positive results also emerged from EMBRACE, a two-part, Phase 2 safety study testing nusinersen in 21 children between seven and 49 months old who had SMA. EMBRACE accepted children who were excluded from earlier nusinersen trials for a variety of reasons, including age at symptom onset and how many copies of mutant SMN2 they carried. Because the gene is highly prone to genetic duplication, SMN2 copy number can be as high as four, and, in general, more copies means milder symptoms. Children who completed the 14-month blinded period could go on to the 30-month, open-label part 2 of the study. Primary outcomes were safety and tolerability, with secondary objectives being pharmacokinetics.
PerryShieh from the University of California, Los Angeles, presented safety data from the first part of EMBRACE. “[Nusinersen] demonstrated a favorable safety profile with no new or unexpected safety signs,” he said. Five children in the treatment arm and three in the placebo had serious adverse events, but none were thought to be related to the treatment. Also, exploratory measures of motor function hinted at improvement. Seven of nine children who had symptoms of SMA before they were seven months old improved in more HINE categories than they worsened in. In contrast, none of the four children with SMA type 1 who were on placebo responded. In eight children with later-onset disease, those taking nusinersen scored better on the HINE, with a mean improvement of approximately five points on day 302, compared with zero for those on placebo. However, two of three children in the placebo group also qualified as responders, suggesting that improvements may be unrelated to drug or the criteria for responder is not stringent enough. Twenty children have now enrolled in EMBRACE part 2.
Results from an even smaller cohort of children with SMA type 2 or 3 who participated in a clinical trial and its open-label extension also hinted at nusinersen benefits. These diseases are milder and start later than type 1 SMA, and patients usually have three or more good copies of SMN2. Children with SMA 3 can stand and walk, although some may require aids, while those with SMA 2 are usually unable to do either independently. Jacqueline Montes, Columbia University, New York, presented data on the 14 children who could walk during the CS2 or CS12 trials and had performed a six-minute walk test. The group included kids aged 2 to 15, 13 of whom had SMA 3 and one who had SMA 2. Except for one child, all were able to walk independently. Montes reported that these children walked a median of 17 additional meters at day 253, and 98 meters farther than a baseline median of 251 meters at day 1,050 of treatment.
Attendees also learned how patients on nusinersen are faring in private practice. Physicians began treating patients with nusinersen even before it was on the market because Biogen’s “expanded access program” allowed them to request the drug for patients not eligible or able to participate in trials. “We’re starting to get real-world data, including some from older patients with a variety of symptoms,” said Finkel. He and Day presented case studies illustrating the conundrum of who to treat, when, and how. For example, a 10-year-old girl with SMA type 2 started taking nusinersen and noticed improvement, which waned toward the end of the four-month period between doses. “Should we treat her every three months?” asked Finkel. “That’s a hard sell for insurance companies that are struggling to cover the cost of dosing every four months,” he said.
Also, what about patients who have had their spines surgically fused and cannot receive nusinersen via lumbar injections? Data from Aravindhan Veerapandiyan, University of Rochester Medical Center, New York, suggested cervical puncture could work as an alternative. He succeeded in delivering five standard 12 mg/5 ml doses of nusinersen into the CSF of three patients in this way. However, as Finkel noted, it is not yet known if this route of administration will be effective. Others wrestled with whether there should be an age or stage limit for treatment. “Is there a population for whom it makes no sense to use this expensive drug?” asked Day, describing a nearly paralyzed man with SMA2 who gained mobility of a finger and reported improved stamina and a stronger voice after treatment. On the other end of the spectrum, clinicians are grappling with whether patients with mild or nonexistent symptoms should be treated. For example, a toddler who carries homozygous SMN1 mutations and four good copies of the SMN2 gene scored above average in motor tests, but is steadily losing her edge. Day asked, should she be treated?
Familial Amyloid Polyneuropathy
In contrast to type 1 SMA, familial amyloid polyneuropathy (FAP) is a rare, slowly progressing degenerative condition caused by accumulation of amyloid fibrils of transthyretin in a multitude of tissues, including peripheral nerves and the heart. Patients in stage 1 have difficulty walking, and by stage 2 can no longer walk unassisted, are in pain, and may have other symptoms, from diarrhea to heart problems. When the latter predominates, the disease is referred to as familial amyloid cardiomyopathy, or FAC. Because amyloid deposits build up slowly, symptoms appear any time from early adulthood to old age, but progression from stage 1 to 2 takes only a couple of years.
Ionis developed inotersen to knock down transthyretin transcripts, and last year reported that this ASO halted progression of FAP. At AAN, John Berk, Boston University, summarized data from 172 adult participants with FAP stage 1 or 2 in the NEURO-TTR Phase 3 trial. This 15-month trial assessed efficacy and safety of weekly subcutaneous injections of 300 mg inotersen (May 2017 news). Participants were randomized 2:1 to treatment and placebo, and assessed for change on their baseline score on the Norfolk Quality of Life Diabetic Neuropathy (Norfolk QOL-DN) scale, which measures the impact on patients’ lives caused by nerve fiber degeneration, and on the modified neuropathy impairment score +7 (mNIS+7), which measures nerve function, muscle strength, and pain. The Norfolk QOL-DN ranges from 1–136 points, the mNIS+7 from 1–346; on both, higher scores are worse. In the treatment arm, 37 percent of people improved over baseline in the mNIS+7, and 50 percent improved on the Norfolk QOL-DN. How about the placebos? This is only meaningful compared to them. Eighty-one percent of participants completed the trial, and of those, 95 percent chose to continue receiving treatment in an ongoing open-label extension.
All the same, Berk noted some serious safety issues. Three patients suffered severe kidney malfunction. Three patients had thrombocytopenia, i.e., extremely low platelet counts, believed to be related to the drug. Even worse, one died from a subsequent intracerebral hemorrhage. The two others recovered after stopping the ASO and taking corticosteroids. Berk noted that the patient who died had received inotersen for nine weeks with no monitoring of platelet count. “The reason for the precipitous drop in platelets remains unclear,” he said. No additional serious cases of thrombocytopenia have developed since then, Berk said, suggesting that with proper monitoring, platelet numbers can be controlled.
Thomas Brannagan, Columbia University, New York, said at AAN that with weekly monitoring in the extension study, ASO dosing is now being stopped or reduced if platelets drop below a set threshold. Similarly, kidney problems appear to be manageable with routine testing. Nevertheless, three patients in the extension suffered serious adverse events considered to be related to treatment. Brannagan did not say what these events were.
His poster detailed interim efficacy results from the extension as of September 15, 2017. The study included 134 patients with a mean age of 64 years, of whom 85 had been on inotersen and 49 on placebo. Among the former, mNIS+7 scores inched up nine points; that is 41 points less than projected based on progression in the placebo group (image below). Similarly, the mean Norfolk QOL-DN scores worsened by four points, about 17 points less than placebo projection. Patients who switched from placebo to inotersen also benefitted, but their gains were more modest, with mNIS+7 scores worsening by 31 points on average, and Norfolk QOL-DN scores by 11 points. “The impression is that the earlier the intervention, the greater the chance of improvement,” said Berk. However, most of the improvement occurred before the open-label extension. During the extension, people who had been on inotersen continuously seemed to plateau, or even begin to deteriorate again, indicating the drug seems to delay, but not stop, the disease process. Inotersen is now under priority review for FDA approval.
Antisense Against Amyloidosis. People with transthyretin amyloidosis who took inotersen continuously scored better (blue) on neurological (left) and quality of life (right) tests than those on placebo (red). After switching to inotersen they did better than projected (dashed line). [Courtesy of John Berk, Boston University.]
Alnylam Pharmaceuticals in Cambridge, Massachusetts, has adopted a different strategy to knock down transthyretin. Patisiran, a small RNA encapsulated in a lipid nanoparticle, destroys transthyretin mRNA by engaging the gene-silencing complex RISC. At AAN, David Adams,National Reference Centre for FAP INSERM in Paris, summarized results from the Phase 3 APOLLO trial (Adams et al., 2017). Patients in APOLLO had FAP caused by a variety of TTR mutations. Most had heart damage, including thickened ventricle walls. They received intravenous injections of 0.3 mg/Kg patisiran every three weeks.
More than half of the 225 participants on patisiran, versus only 4 percent on placebo, registered drops on the mNIMS+7 after baseline. The improvements surfaced nine months into treatment, and average scores continued to improve until the end of the study at 18 months. Adams said the results suggested reversal of disease. A press release from Alnylam additionally claims a positive correlation between TTR knockdown and improvement in neurologic function. These data were presented at the 16th International Symposium on Amyloidosis, held this past March in Kumamoto, Japan. Secondary outcome measures, including Norfolk QOL-DN scores and gait speed, also reportedly improved on patisiran.
The treatment appeared safe. Adams said the frequency of serious adverse events was similar in the groups, with deaths in the placebo arm occurring at 7.8 percent versus 4.7 percent in the treatment arm. There were no signs of liver or kidney problems, nor thrombocytopenia. The majority of adverse events were mild to moderate peripheral swelling or reactions at the injections site, which rarely caused patients to stop treatment.
Adams reported an association between patisiran treatment and reduced thickness of the ventricular wall in a subgroup of 126 patients who had abnormally large left ventricular walls at baseline. The patients’ heart functions improved, and they had lower levels of a cardiac stress biomarker called N-terminal pro b-type natriuretic peptide in the blood.
How do inotersen and patisiran compare? Their trials were not run side by side, used different endpoints and methodologies, and recruited patients with different characteristics. Different safety concerns have arisen for each of the drugs, although as of now they both appear largely safe and well-tolerated. Whereas inotersen is delivered subcutaneously, patisiran requires intravenous injection. Alnylam is developing a new RNAi therapeutic that can be delivered subcutaneously, which Adams said potently and durably reduced TTR in Phase 1.—Marina Chicurel
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC, ENDEAR Study Group.
Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy.
N Engl J Med. 2017 Nov 2;377(18):1723-1732.
PubMed.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, Schneider E, De Vivo DC, Finkel RS, CHERISH Study Group.
Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy.
N Engl J Med. 2018 Feb 15;378(7):625-635.
PubMed.
At AAN, Sights Set on Antisense Therapies for Diseases of the Brain
In recent years, two antisense oligonucleotide therapies have hit the market, at least nine more are in clinical trials for the treatment of neurologic diseases, and nearly two dozen are in preclinical testing, including ASOs for Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis, frontotemporal dementia, spinocerebellar ataxias, pain, and rare disorders. At the 70th annual meeting of the American Academy of Neurology, April 21 to 27 in Los Angeles, researchers reported on Phase 3 data and open-label extensions for treatment of spinocerebellar ataxia and familial amyloid polyneuropathy (see Part 1 of this story) and on the latest data in their efforts to develop therapeutic ASOs for suppressing expression of huntingtin, tau, and C9ORF72 transcripts.
“Antisense oligonucleotides are bringing a new wave of enthusiasm and excitement to the field of neurologic diseases,” said John Day of Stanford University.
Until recently, researchers doubted it was feasible to get enough ASOs across the blood-brain barrier and distributed across the human cortex, or subcortical regions, for them to effect a therapeutic benefit. However, evidence has been accumulating that this problem is surmountable. For example, at AAN, Frank Bennett from Ionis Pharmaceuticals in Carlsbad, California, showed that after injecting an ASO against mRNA for the transcription factor MALAT1 into the CSF of nonhuman primates, levels of the target RNA dropped in every one of more than 30 different brain regions analyzed. Reductions ranged from 20 percent in deep areas, such as the putamen and globus pallidus, to more than 90 percent in the spinal cord and several areas of the cerebral and cerebellar cortices. MALAT1 expression in most brain regions fell to or below half. Bennett noted that increasing the potency of the ASO knocked down MALAT1 transcripts more efficiently in deep brain structures. Similarly, in an online presentation last month, Michael Ehlers of Biogen, Cambridge, Massachusetts, claimed that in a nonhuman primate, an intrathecal bolus of an anti-α-synuclein ASO robustly lowered both α-synuclein mRNA and protein, not only in temporal and dorsal prefrontal cortices, but in the substantia nigra, an important target for Parkinson’s treatment.
In the Clinic. Some antisense drugs for neurological diseases are approved, others are in clinical trials. [Courtesy of Frank Bennett, Ionis Pharmaceuticals.]
At AAN, Sarah Tabrizi, University College London, summarized data from a Phase 1/2a trial of the anti-huntingtin ASO IONIS-HTTRx. Last March at the CHDI Huntington’s disease conference, Tabrizi reported that the treatment lowered mutant huntingtin in the CSF by 40–60 percent during the 13-week trial (Mar 2018 news). In Los Angeles, she added data from the follow-up period when participants were off the drug. She said that four months after treatment had ended, participants in the treatment group scored no better than those on placebo in the UHDRS, a composite scale of HD severity based on measures of motor, cognitive, and behavioral health. “We don’t expect clinical change over seven months in a study with a relatively small number of patients in stage 1 HD,” Tabrizi said.
In post hoc analysis, the researchers did find some associations between reduction in mutant huntingtin in CSF, improved scores on the UHDRS, and individual components of the composite at day 85, the last day of dosing. However, no correlation was found with the Stroop test, a measure of attention and mental flexibility, nor with total functional capacity, an indicator of a person’s ability to live independently. “These findings are post hoc and exploratory; they require replication and extension in a longer and larger study,” cautioned Tabrizi.
Ionis’ Laurence Mignon described a clinical trial to test IONIS-MAPTRx, an ASO that destroys tau pre-mRNA. This ASO recruits RNAse H to exon 9 of the tau transcript. The primary goal of the trial is to assess safety and tolerability in patients with mild AD, but it will also examine CSF pharmacokinetics as a secondary endpoint, as well as biomarkers and clinical outcomes as exploratory measures. Completing the first part of a plan to test multiple ascending doses, a group of eight patients, aged 50–75 years, recently received the last of four intrathecal injections of the ASO, with the second group just getting started, said Mignon. The researchers expect to test four cohorts, totaling 44 patients. Tim Miller and colleagues at Washington University, St. Louis, had previously reported that the first-generation tau ASO, TauASO-12, prevented and even reversed tau pathology when injected into the spinal fluid of transgenic mice expressing the human tau gene. The ASO also normalized nest-building behavior, and lengthened lifespan (Jan 2017 news). In Los Angeles, Mignon also pointed to studies indicating that cell and animal models tolerated tau reduction well, especially in adulthood. Moreover, intrathecal injections of IONIS-MAPTRx in cynomolgus monkeys knocked down tau mRNA by more than half in the spinal cord, frontal cortex, and hippocampus, she said.
Robert Brown, University of Massachusetts in Worcester, expects to move ASOs targeting RNAs implicated in ALS into the clinic by the end of the year. The most common genetic cause of familial ALS and frontotemporal dementia (FTD) is a large G4C2 hexanucleotide repeat expansion in the first intron of the C9ORF72 gene. Wave Life Sciences, an oligonucleotide therapies company based Cambridge, Massachusetts, has created the ASO WVE-3972-01 to silence the expanded allele. Unlike other ASOs, which are made as a mixture of stereoisomers, WVE-3972-01 is stereochemically pure. Although all molecules of a given ASO share the same nucleotide sequence, stereoisomers differ in the three-dimensional orientation of atoms around some of their bonds. This means the isomer with the most therapeutically useful configuration can get diluted by others in the mix whose shape is suboptimal. By controlling the geometry of the ASO backbone during synthesis, Wave Life Sciences produces uniform oligonucleotides that can be selected for better RNAse H targeting and resistance to degradation by single-strand nucleases (Iwamoto et al., 2017). Brown reported that in head-to-head comparisons, stereopure ASOs were more selective and more potent than regular ASOs with mixed backbone configurations.
So what about WVE-3972-01? When injected into the cerebral ventricles of mice expressing the human C9 gene with a repeat expansion, the ASO found its way into the cortex and spinal cord, and was still detected eight weeks later in the nuclei of spinal cord motor neurons. At that time, levels of toxic poly-glycine-proline (GP) peptides encoded by the mutant C9 gene dropped by more than 80 percent and toxic RNA foci were cut roughly in half, while total C9 mRNA and protein levels fell only slightly. Brown said his group is now putting WVE-3972-01 through toxicology tests.
In response to an audience question, Brown acknowledged that C9 produces sense and antisense mRNA transcripts that both encode toxic poly-dipeptides; however, WVE-3972-01 only targets the sense transcript. “For me, the $64,000 question is: Are we going to have to reduce both strands?” he said. Brown’s data suggest that at least polyGP, which can also be made from the antisense strand, is robustly lowered by an ASO that targets only the sense strand. However, this ASO leaves the proline-arginine dipeptide, encoded by the antisense strand, unchanged.
Lindsey Hayes of Johns Hopkins University in Baltimore showed data hinting that targeting either one of the strands alone may be enough to combat at least some aspects of ALS pathology. Using induced pluripotent stem cells derived from C9 patients, Hayes found that treating the cells with ASOs against either of the two strands restored nuclear pore proteins and Ran, a nuclear protein required for nucleocytoplasmic transport, to their normal locations within cells. “Surprisingly, both [ASOs] independently rescue these pathologies,” said Hayes. “In vitro, dual therapy does not appear to be required to rescue nuclear transport.”
What Next?
On April 20, Biogen announced a $1 billion deal with Ionis to develop ASOs to treat neurologic diseases, including AD, PD, ALS, FTD, and progressive supranuclear palsy. Both companies hope to replicate the success of nusinersen. Ionis will handle early stages of target validation, Biogen will take over development from toxicology to commercialization. In an investor presentation, Biogen’s Ehlers said he expects to have up to seven candidates in the clinic within two years and to validate 50 targets in the next decade. BIIB5Rx, IONIS-BIIB6Rx, and IONIS-BIIB7Rx are in preclinical development for undisclosed neurodegenerative diseases. “We think ASOs are the most advanced genetically based approach for targeting neurological diseases,” said Ehlers, adding he expects ASO validation and production to be faster than for small molecules.
Scientists at AAN also discussed other gene therapies (see Part 1 of this story). Receiving intrathecal doses of an expensive ASO drug for life has obvious disadvantages, but it avoids irreversible changes that might come with permanently modifying genes and potentially harmful immune reactions elicited by some gene delivery vectors. Ehlers considers ASO and gene therapy complementary and said Biogen is exploring combination approaches. Others worried that combining ASO drugs with gene therapy would raise enormous price tags even further. Nusinersen, the ASO approved for treating spinal muscular atrophy, at present costs $750,000 for the first year of treatment and $375,000 each year thereafter.—Marina Chicurel
New Alzheimer’s and Parkinson’s Immunotherapy Data at AAN
For neurodegeneration researchers, highlights at the 2018 conference of the American Academy of Neurology, held April 21–27 in Los Angeles, California, were centiloid analysis of PET scans, the latest cognition update from people treated with the anti-Aβ antibody aducanumab, as well as new data from Parkinson’s patients on an anti-α-synuclein antibody. “The inclusion of results in centiloids is an important development,” Chris Rowe, University of Melbourne, Australia, wrote to Alzforum. “It will allow a more meaningful comparison of the cohorts’ baseline amyloid burden, and the effectiveness of amyloid clearance, [with those measures] in other drug trials, something that is otherwise limited by the use of different amyloid tracers and analysis methods. This is exactly why the centiloid method was developed.”
Opening the first session on clinical trials in neurodegenerative diseases, Samantha Budd Haeberlein, Biogen, Cambridge, Massachusetts, presented 24- and 36-month data from the ongoing, long-term extension of the Phase 1b PRIME trial. PRIME tests the safety, tolerability, pharmacodynamics, and pharmacokinetics of aducanumab in people with prodromal and mild Alzheimer’s disease. As reported at CTAD last year, by 18 months amyloid deposition in people receiving the highest dose of aducanumab, 10 mg/kg, had dropped below the preset threshold for amyloid positivity—in this case a whole-brain standardized uptake value ratio (SUVR) of 1.1 for florbetapir PET (Dec 2017 conference news).
At ANN, Budd Haeberlein reported the PET data fitted to the Centiloid scale, which was designed to harmonize PET data across different tracers and detection systems (Klunk et al., 2015; Nov 2014 news; Feb 2013 news). “Different scanners, tracers, and even analysis methods contribute to the absolute numbers we see with SUVRs,” she said. “Essentially, the Centiloid scale is an attempt to standardize quantitative measures of PET imaging.” Other groups are adopting the scale, including ADNI-3 (Weiner et al., 2017; Jack et al., 2017; Yun et al., 2017).
Say It in Centiloids. Brain amyloid deposition fell by 70 centiloids over 36 months in people with mild or prodromal AD who were treated with aducanumab. [Courtesy of Biogen.]
As expected, the shape of the Centiloid and SUVR graphs look the same, with florbetapir binding falling in an aducanumab dose- and time-dependent manner. “So what have they gained by the conversion to centiloids?” asked the scale’s creator, William Klunk, University of Pittsburgh. “Now the absolute value of the decrease holds some meaning that is generalizable to past or future studies,” he wrote to Alzforum.
In PRIME, amyloid continued to fall up to 110 weeks in people on the 6 mg/kg and 10 mg/kg doses, then leveled off by week 166 at a reduction of about 70 centiloids below baseline for both doses. People on 3 mg/kg tended to keep losing amyloid to week 166, as did those who started the trial taking 1 or 3 mg/kg or placebo, but switched to 3 or 6 mg/kg after the 12-month placebo-controlled period.
Rowe and Klunk were impressed with the 60-70 centiloid reduction. Klunk thinks the data imply a high level of target engagement. Whereas the key question had been whether anti-Aβ antibodies could meaningfully reduce amyloid deposition, now researchers can ask if amyloid is the right target, if treated early enough, he wrote.
Exploratory clinical data hint that, possibly, it might. Certainly that’s the big hope. Budd Haeberlein presented new data suggesting a continued benefit of aducanumab over time on the Mini-Mental State Exam (MMSE) and the clinical dementia rating sum of boxes (CDR-SB) scales (image below). The antibody slowed decline, suggesting it may have modified the disease process, but did not halt it. The effect was dose-dependent with the curious exception of the 6 mg/kg dose, which has been out of synch with the 3 mg/kg and 10 mg/kg doses since the early days of the trial. Budd Haeberlein did not explain this anomaly, but instead emphasized that the sample size is small, with only 166 people dosed in the first, placebo-controlled year of the study, and 117 continuing in the long-term extension. Other researchers have noted that the 6 mg/kg group began treatment at a different time than the 1, 3, and 10 mg/kg group.
Did Cognitive Decline Slow? In general, people on aducanumab had better CDR-SB (top) and MMSE (bottom) scores than those on placebo. [Courtesy of Biogen.]
Gil Rabinovici, University of California in San Francisco, complimented the work. He asked about the relationship between the Aβ-PET reduction and the change in clinical outcomes. Did people with more amyloid removal do better? Budd Haeberlein responded that in 2016, Biogen observed that people who had a reduction of 0.15 SUVR units or more indeed were more likely to benefit clinically, but the company has not yet updated the analysis.
Summarizing the latest data on amyloid-related imaging abnormalities, Budd Haeberlein said ARIA usually occurred early in the course of treatment, was typically mild and asymptomatic, and tended to resolve or stabilize within four to five weeks. She concluded that, altogether, the PRIME data support further investigation of aducanumab in the ongoing Phase 3 EMERGE and ENGAGE trials.
On to Parkinson’s
In Los Angeles, Biogen also presented new clinical data on BIIB054, a monoclonal antibody that binds pathological, aggregated α-synuclein. Miroslaw Brys said that BIIB054 is approximately 400 times more selective for aggregated α-synuclein than for the monomeric form of the protein. At the AD/PD conference in Vienna, Biogen reported on the safety, tolerability, and pharmacokinetics of BIIB054 in healthy volunteers (May 2017 conference news). At AAN, Brys presented the first results of a cohort with Parkinson’s disease (PD) patients in the same Phase 1 clinical trial.
Thirteen men and five women with early PD, aged 47–75, received a single intravenous dose of either 0, 15, or 45 mg/kg of BIIB054. Nine were on no PD medication, five were on levodopa, two on rasagiline, and two on both rasagiline and levodopa. Over the next 16 weeks, participants underwent clinical assessments, two MRI scans, and had blood and cerebrospinal fluid (CSF) drawn to track BIIB054 levels and immune response to the antibody.
Brys reported that the pharmacokinetics of the antibody in patients were similar to those in healthy volunteers—it had a 33-day half-life in serum, reached a plasma concentration approximately threefold higher in the high- than in the low-dose group, while only 0.4 and 0.3 percent found its way into the CSF in the high- and low-dose groups, respectively. None of the patients developed anti-BIIB054 antibodies.
Brys said that as in healthy volunteers, BIIB054 formed complexes with α-synuclein in plasma. That both doses formed similar amounts of complex suggested near-complete saturation of blood synuclein with antibody. Attendees wondered why the antibody bound so much plasma synuclein if its affinity for monomers was so low. Brys explained that, although BIIB054 is more selective for aggregated than soluble α-synuclein, at high doses it is expected to bind the soluble form. Brys said there were no serious adverse events, and no obvious worsening of PD symptoms in either placebo or treatment arms during this short trial. The SPARK Phase 2 study of BIIB054 is enrolling now.—Marina Chicurel






Comments
No Available Comments
Make a Comment
To make a comment you must login or register.