At the third annual Zilkha Symposium, held April 15, 2016, in Los Angeles, scientists from the United States and Europe wrestled with the immense complexity and heterogeneity of Alzheimer’s disease. The program ranged from genetics to clinical symptoms to drug targets. Researchers discussed how genes point to amyloid, tau, and the immune system. They debated the merits of diagnosing Alzheimer’s disease based on symptoms versus biomarkers, and considered a panoply of medications now in trials. Rather than being overwhelmed by complexity, however, researchers were confident that even attacking one of multiple pathogenic pathways in Alzheimer’s could help people with the disease, perhaps soon. Highlights included new approaches to Aβ oligomer morphology, the interplay between amyloid and microbes, and the importance of the brain’s vascular system. Read Amber Dance’s series.
Aβ Oligomers Purified from Human Brain
Scientists and clinicians gathered at the University of Southern California in Los Angeles on April 15, 2016, for a day jam-packed with a dozen presentations. It was the third annual Zilkha Symposium on Alzheimer Disease & Related Disorders, co-organized by USC’s Berislav Zlokovic, David Holtzman of Washington University, St. Louis, and Rudy Tanzi of Massachusetts General Hospital in Boston. In talks that stretched from genetics to clinical symptoms to therapeutic targets, the overarching theme was coming to grips with complexity and heterogeneity. Scientists agreed they need to better understand not only Alzheimer’s central pathways of Aβ and tau, but also the many additional processes that influence its pathogenesis in important ways, such as neuroimmune and neurovascular regulation. On the Aβ topic, speaker David Brody of Washington University in St. Louis offered preliminary results that might open a new door to the study of oligomers. Brody purified oligomers from human brain and came up with a set that look nothing like the oligomers and fibrils previously obtained by scientists working with mouse models or synthesizing oligomers in vitro.
Zilkha Neurogenetic Institute
[Keck School of Medicine Public Relations]
Scientists know that while the distribution of amyloid plaques does not correlate tightly with dementia in people, oligomeric Aβ does suppress learning and memory, and kills neurons in various experimental systems. However, for years, the individual labs studying Aβ oligomers have seemed to find, or make, slightly different types of oligomer. Scientists have isolated dimers from human cerebrospinal fluid and cultured cell lines, collected dodecamers from the brains of AD models, or synthesized their own artificial versions in vitro. Only some oligomers are toxic at low concentrations, and labs have not broadly reproduced each other’s methods and findings, leaving the field at an impasse (see Oct 2011 webinar). What’s more, scientists have cautioned that studying oligomers in the lab may alter their properties. Detergents used in purification protocols or gel electrophoresis buffers can induce oligomerization of monomers within minutes (Hepler et al., 2006; Watt et al., 2013) Roughly douncing or otherwise homogenizing tissues might break up larger aggregates, Brody speculated. “It is not clear what exactly is going on with amyloid-β oligomers, and which ones are most toxic,” Brody said.
Zlokovic called Brody’s work “the most detailed and comprehensive analysis of oligomers from brain tissue that I saw so far.” However, Brody noted his results are still preliminary. “I do not think we have in any way solved this problem; what we are doing is pushing the technical envelope a little bit.”
Brody, and Thomas Esparza in his lab, set out to purify native Aβ oligomers from human brain tissue of people who died of AD, despite warnings from colleagues. “Many people tried and gave up,” Brody said. “I have been told a dozen times not to do this, that this was impossible.” Part of the challenge is that oligomers are thought to occur in minute concentrations in the brain; another was the lack of a good assay to quantify the presence of oligomers throughout the steps of a purification protocol.
Brody and Esparza thought they could succeed because they had previously developed a quantitative ELISA for oligomers (see Feb 2013 news). Another advantage, Brody told Alzforum, was the availability of relatively large quantities of AD brain tissue from the Washington University Alzheimer’s Disease Research Center. Other scientists trying to develop protocols for purifying amyloid oligomers have had to rely on non-human sources, Brody said, such as synthetic oligomers that might differ from the natural versions. In contrast, Esparza was able to use human tissue for all his experiments, over years spent developing the protocol.
The researchers started with amyloid-loaded brain tissue from people who had died of autopsy-confirmed Alzheimer’s. Esparza minced cortical tissue, then homogenized it in buffer with CHAPS, a detergent he found does not facilitate Aβ oligomerization. He spun the homogenate in a centrifuge to pellet out large debris, including plaques. Then he took that supernatant, which included a mix of monomers and oligomers, and spun it again at 475,000 x g. Esparza had to use a special ultracentrifuge to reach this enormous speed. He included a sucrose cushion at the bottom of his tubes to catch the oligomers; otherwise, they would become irretrievably stuck to the tube walls, Brody said. Monomers remained in the supernatant.
Next, Esparza limited his preparation to oligomer size with size-exclusion chromatography, and immunoprecipitated the oligomers with two different antibodies to Aβ. All the while, he checked his intermediate preps to make sure the oligomers were still present. One important trick, Brody said, was to coat every pipette tip, test tube, or other apparatus with albumin. Otherwise, the Aβ sticks to the equipment and a bit gets left behind at every step.
As it turned out, it was not impossible to isolate oligomers from brain—just “very, very difficult,” Brody said. Overall, Esparza lost less than 30 percent of the amyloid he had in the original homogenate, and he concentrated the protein by 10,000 times. The oligomers’ structures remained stable throughout the procedure, Esparza believes, because they had the same size according to size-exclusion chromatography in the initial lysates and final purified preparation. No new oligomers formed; Esparza confirmed this by spiking in monomers, which stayed monomers.
What kinds of oligomer were in the AD brains? Some were large, more than 500 kilodaltons according to size-exclusion chromatography. Using an electron microscope, Esparza spied clusters of spheres, each 10-20 nanometers across. Some clusters contained just three or four spheres; others grouped together dozens. They were decidedly non-fibrillar. “What we see does not look like anything that has been previously reported,” said Brody. He suspects each sphere contains one or more Aβ molecules, plus associated proteins the amyloid may bind.
Norelle Wildburger in Brody’s lab used mass spectrometry to identify the specific Aβ species in his oligomers. She saw an assortment. Besides those that start with Aβ’s first amino acid, there were others truncated at various sites up to position 11. Measuring Aβ’s other end, she saw peptides ending between positions 34 and 43. The molecules were sometimes modified, for example having lost amine groups or gained cyclized pyroglutamate.
Brody suspects, and other scientists at the Zilkha Symposium agreed, that some previous studies of Aβ oligomers may have been confounded by their protocols. Maria Carrillo of the Alzheimer’s Association said it was intriguing to consider oligomers might be larger than previously thought, and that some other oligomer studies might have focused on artefacts. However, Brody cautioned that his own prep still might be subject to other types of artefact. For example, oligomers might form postmortem as tissues cool from body to room temperature, before the medical examiner arrives.
Ideally, one would like to detect oligomers in the living brain, perhaps with imaging agents specific to oligomer epitopes. Alas, Brody said such tracers are nowhere near ready (Feb 2016 news; Dec 2014 news). Scientists are also making progress with methods to detect oligomers in cerebrospinal fluid, but still wish for more sensitivity in those assays (see Jul 2012 conference news; Feb 2014 news).
What does seeing a zoo of larger Aβ oligomer species in human AD brain mean for the field? “It is, in one sense, a giant step backwards, because a lot of what we thought we knew about Aβ oligomers may not be that relevant to the human brain,” Brody said. Potentially, therapeutics that target synthetic oligomers might not work in people, he speculated. However, he added, his work also offers ways to take oligomer research forward. “It is, perhaps, an opportunity to look more broadly at the other forms of amyloid-β oligomers that might exist in human brain,” Brody told Alzforum. Once published, Brody will make the detailed protocol publicly available.
Many questions arise from this ongoing work, Brody noted. For one, he is interested in comparing the various Aβ constituents and their proportions between human and mouse brains. For another, he wants to test how the oligomers interact with plaques, and if plaques from the brains of donors who died without dementia are better at buffering loose oligomers. This might explain in part why people with brain amyloid were protected from dementia.
Sangram Sisodia of the University of Chicago called Brody’s presentation “the coolest stuff I saw.” He said it would be key to show which species are toxic. Brody plans to inject mouse brains and check for synaptic degeneration or loss. He also wants to expose hippocampal slices to the oligomers and measure effects on long-term potentiation. Washington University’s Holtzman said the work took a different direction from the rest of the field, and wondered if these oligomers might be the long-sought connection between Aβ and tau—in other words, if their presence drives the emergence of tauopathy.—Amber Dance
Microbial Hypotheses Intrigue at Zilkha Alzheimer’s Meeting
Microbes both friend and foe might contribute to the formation of amyloid plaques, according to researchers who spoke at the third annual Zilkha Symposium on Alzheimer Disease & Related Disorders, held April 15, 2016, in Los Angeles. Rudy Tanzi of Massachusetts General Hospital in Charlestown presented ongoing research on his and Robert Moir’s hypothesis that Aβ is an antimicrobial peptide, reporting that Salmonella bacteria can seed formation of plaques in the brains of mice. In turn, Sangram Sisodia of the University of Chicago focused on the bacteria that normally populate the gut. Shifting that teeming mass with intense antibiotic treatment reduced plaque burden in mice by half, he told attendees.
Can Salmonella seed amyloid plaques? [Courtesy of NIAID.]
Making the case for a role for pathogens in AD, Tanzi noted that many of the genes recently found to be involved in Alzheimer’s disease risk function in the immune system. For example, high levels of TREM2 promote phagocytosis by microglia, while mutations in CD33 reduce phagocytosis rates, increasing amyloid plaque burden and overall Aβ load, Tanzi said.
Tanzi was the first to say his hypothesis warrants healthy skepticism. Referencing the Schopenhauer quote of the three stages of truth, whereby a novel idea is first ridiculed, then violently opposed, then finally accepted as self-evident, Tanzi placed his assertion that Aβ is an antimicrobial peptide between stages one and two. Meeting attendees were diplomatic, telling Alzforum the idea was intriguing and worthy of further investigation.
Anti-Microbial Aβ?
Antimicrobial peptides (AMPs) are charged peptides of 12-50 amino acids. “They are our first line of defense,” Tanzi said, explaining that these little peptides oligomerize into a “nanonet” of fibrils that trap an invading pathogen before the more differentiated adaptive immune system goes after it. In fact, several amyloid-forming proteins have antimicrobial properties (reviewed in Kagan et al., 2012). The similarities between AMPs and neuropeptides such as Aβ have led some scientists to hypothesize that neuropeptides might have anti-infective functions (Schluesener et al., 2012).
Previously, Tanzi’s group, together with the Moir laboratory at MGH, had proposed that Aβ counters microbes in vitro, and reported that amyloid-containing brain homogenates from people who died of Alzheimer’s have high antimicrobial activity (Apr 2009 conference news; Mar 2010 news). At the Zilkha meeting, Tanzi provided an update of the MGH scientists’ effort to explore the AMP hypothesis in a range of model systems.
They started small with human neuroglioma cultures that the researchers infected with Candida albicans, a type of yeast. Overexpressing Aβ in these cells protected them, doubling the number that remained uninfected, Tanzi told the audience. The researchers next infected Caenorhabditis elegans with Candida. In unprotected worms, the fungus grows to the point of breaking through their bellies. “It looks like the movie ‘Alien,’” Tanzi said. Half of the nematodes died within three days, but expressing Aβ in their muscle cells protected them; more than half were still alive after six days. Next Tanzi and Moir turned their sights on fruit flies, infecting Drosophila with Candida. Once again, wild-type flies perished within 50 days but those overexpressing Aß made it to 60.
Finally, the authors used Salmonella to infect the brains of four-week-old mice. For wild-type mice, this type of bacteria-derived meningitis means death within 60-72 days. Mice lacking the APP gene succumbed even faster, by 54 days, but 5xFAD mice survived to a maximum of 96 days, as if the excess Aβ protected them.
What blew the researchers’ minds, Tanzi said, was finding amyloid plaques when they examined the brains of the 5xFAD mice as soon as 48 hours after injecting Salmonella. Normally, these mice do not have plaques at the young age used in this study, but the infected animals did. And in the middle of each plaque was a Salmonella bacterium—the microbe seeded plaque formation around it. (Alzforum first reported this result in Apr 2015 conference news.)
This is what one would expect if the Aβ were an antimicrobial peptide. Moir and Tanzi have proposed a model they call the anti-microbial protection hypothesis, by which a subclinical or asymptomatic infection might seed amyloid formation, kicking off Alzheimer’s disease as a form of collateral damage. Older people might be more susceptible to this process as pathogens sneak into the brain through a blood-brain barrier compromised by age and adaptive immunity begins to wane (see Part 3 of this series).
Some other labs are starting to back up this hypothesis, reporting that Aβ inhibits both influenza virus and herpes simplex virus-1 (HSV1) (White et al., 2014; Bourgade et al., 2015; Bourgade et al., 2016). One implication, Tanzi told Alzforum, is that Aβ-lowering therapies should perhaps not be too potent lest recipients lose some innate protection against infectious agents. “Amyloid is not just junk,” he told the Zilkha audience. “We want to reduce it but not wipe it out.”
Microbiome Meets Aβ
While Tanzi considered invading microbes, Sisodia’s lab focused on the astounding 100 trillion or so bacteria that naturally inhabit the human gut. There have been previous hints that the natural microbiome could matter to the nervous system. For example, certain gut microbes can produce the neurotransmitter GABA, and germ-free mice have abnormally low expression of brain-derived neurotrophic factor in the hippocampus and cortex, along with abnormal behavior on tests of cognition and anxiety (reviewed in Bhattarcharjee and Lukiw, 2013). The brain’s resident immune cells, microglia, seem to depend on a healthy gut microbiome to mature properly (Jun 2015 news). And a sugar made by Bacteroides fragilis, a common intestinal denizen, protects mice from encephalomyelitis, a model condition for multiple sclerosis (Ochoa-Repáraz et al., 2010).
Moreover, microbes themselves secrete amyloids, for example to help produce a sticky biofilm. These may contribute to neuropathology and AD, scientists posit (reviewed in Hill and Lukiw, 2015). One group found a higher level of infectious burden in people with AD, and proposed that bacterial infection could cause the immune system to confuse its own mitochondria or amyloids for invaders, and thus upregulate inflammation (Bu et al., 2014).
Sisodia hypothesized that the makeup of the intestinal microbiome would influence neuroinflammation, and thus Aβ deposition, in the APPSWE/PSEN1dE9 mouse model of Alzheimer’s. The researchers treated mice from birth with a cocktail of eight antibiotics, assuming this would decrease the number of bacteria in their intestines.
That was not what happened, though. When the scientists checked the mice’s feces and cecum, they found plenty of microbes in both sources. “This came as a bit of a surprise,” Sisodia said. “We thought we would be wiping out the intestinal commensals.”
But were the bacteria that survived the antibiotic onslaught the same as those in untreated mice? Sisodia reasoned that antibiotic-resistant bacteria might have taken over. To gain a bird’s-eye view of the bacterial populations, the authors isolated their 16S rRNA and chopped it up with restriction enzymes to generate restriction fragment length polymorphisms (RFLPs). When they ran these out on a gel, clearly different RFLP patterns emerged between the antibiotic-treated and untreated mice. The populations must differ, Sisodia concluded. The researchers are now performing DNA sequencing to identify the particular bacteria in the mix.
Even if they did not manage to eliminate the microbiome, was just changing it sufficient to alter Aβ in the brain? Indeed it was. Plaque burden was halved in the antibiotic-treated mice. Insoluble Aβ42 levels also dropped, while the amount of soluble Aβ more than doubled.
Thus far in this new research project, the finding is robust only in male mice, Sisodia noted. While there was a trend toward reduced Aβ burden in the females, as well, it stayed below statistical significance with the nine to 10 mice used per group.
Presumably, the gut bacteria altered the peripheral immune response, which in turn changed the biology of the brain. To check immunity in the periphery, the researchers pooled serum from the antibiotic-treated mice and profiled it for cytokines, chemokines, and growth factors. They saw upregulation of several inflammatory mediators in the treated animals, some of which, such as CLC 11, can cross the blood-brain barrier. Sisodia speculated they might enter the brain and activate microglia.
Next, the researchers are raising germ-free 5xFAD mice to check how a missing microbiome affects amyloid deposition. They intend to repopulate those mice with a controlled bacterial population, to test which species or combinations affect amyloid.
Scientists at the Zilkha conference were quite interested in this microbial research direction. “It gives us a whole new perspective to the origin and understanding of AD,” said Philip Scheltens of the VU University Medical Center in Amsterdam. “We have to see how it plays out.”
“We have been scratching the surface of the microbiome, but it is really going to be important,” commented Maria Carrillo of the Alzheimer’s Association. She noted that some scientists have been pursuing a relationship between HSV1 and AD for well over a decade (Jamieson et al., 1991; Feb 2011 webinar). “Perhaps there is something to that,” said Carrillo.
Carrillo is not the only one who thinks so. In a recent editorial in the Journal of Alzheimer’s Disease, 33 AD researchers and clinicians argued that studies of microbes in AD have been neglected (Itzhaki et al., 2016). “We propose that further research on the role of infectious agents in AD causation, including prospective trials of antimicrobials therapy, is now justified,” the authors wrote.
The gut-brain axis is beginning to be explored in neurovascular research, as well. This month, a research collaboration including Costantino Iadecola at Weill Cornell Medical Center in New York reported that changing commensal bacteria with antibiotics reduced ischemic brain injury in mice via changes in T cells trafficking from the intestine to the brain after a stroke (Benakis et al., 2016). For more on neurovascular research in Alzheimer’s, see Part 3 of this series.—Amber Dance
Blood vessels feed the ever-hungry brain, so naturally they have a critical role to play in keeping it healthy or letting it starve. At the third annual Zilkha Symposium on Alzheimer Disease & Related Disorders, held April 15, 2016, at the University of Southern California in Los Angeles, three researchers presented the latest on this topic, which is starting to get traction in AD research. USC’s Berislav Zlokovic, who co-organized the conference, discussed how aging and ApoE4 genotype conspire to cause leaks in the blood-brain barrier. Costantino Iadecola of the Weill Cornell Medical College in New York presented work on how tau blunts the ability of neurons to call in more blood flow when they need additional oxygen. Christer Betsholtz of Uppsala University in Sweden focused on the biology of pericytes, the little contractile cells that envelop blood vessels and manage flow rates.
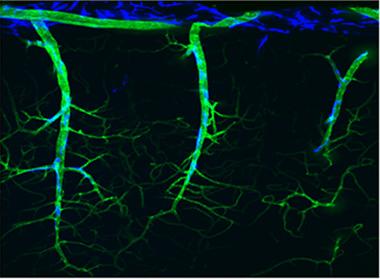
Blood Meets Brain.
Blood vessels (green) dotted by perivascular cells (blue) plunge deep into the cerebral cortex of a mouse. [Courtesy of Laibaik Park and Costantino Iadecola, Weill Cornell Medical College]
The arteries, veins, and capillaries that pervade the brain have started to receive attention from dementia researchers in recent years. “As our knowledge advances, the thinking and focus of the science of cognitive decline has expanded to include blood vessels,” said Roderick Corriveau, a program director at the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. An integrated view of brain function that includes neurons, glia, the vasculature, and immune cells that cross the blood-brain barrier offers a more realistic understanding of neurodegeneration, Corriveau said. In 2014, the National Institutes of Health started using the term “vascular contributions to cognitive impairment and dementia” to categorize such studies. “That was the first recognition of VCID as a scientific field,” Corriveau said. Its scope includes the aging neurovascular unit and its failure to cope with biological insults due to vascular disease, Alzheimer’s biology, metabolic disease, and immune stressors (see Corriveau et al., 2016; Snyder et al., 2014; Apr 2016 conference news).
Tau Acts Alone
Dysfunction of the vasculature might well contribute to the onset or progression of AD, Zlokovic told Alzforum. If so, how might tau interact with the brain’s vasculature? Iadecola’s group has developed a system in which to study blood vessel activity in the mouse whisker barrel cortex. Tickle a whisker, and cortical neurons in its projection area should activate, stimulating more blood delivery. By opening a cranial window above the whisker barrel cortex, the researchers can drip in compounds such as vasodilators or muscle relaxants, and observe how the blood flow changes in different conditions (May 2014 conference news). Scientists already know that Aβ affects diverse aspects of cerebral circulation, Iadecola told Alzforum. It effectively leads to a low-oxygen, high blood-pressure situation in the brain akin to that in a person with hypertension or diabetes.
In the new work, the lab turned its attention to tau. “There is increasing evidence that there is a problem with blood flow in the pure diseases of tau,” Iadecola told Alzforum. For example, a recent study of autopsy brains suggested that tau pathology remodels blood vessel walls (Merlini et al., 2016).
The researchers examined two models of tauopathy, the rTg4510 mouse expressing a repressible tau-P301S; and the PS19 mouse that expresses the same mutant gene under control of the prion promoter, which is active in neurons. Results were similar in both lines. When the scientists tweaked their whiskers, blood flow to the whisker barrel cortex should have risen, but this response was impaired.
Iadecola suspected the problem might be in the activity of NMDA receptors. Upon stimulation, these receptors normally release nitric oxide, which dilates blood vessels. When the researchers inhibited NMDA receptors in the mutant tau mice, manipulation did not affect their response to whisker stimulation, because that pathway was already inactive. “Most likely the coupling between the NMDA receptor and the nitric oxide that is needed to increase flow is not there,” Iadecola said.
In people with Alzheimer’s, the combined actions of Aβ and tau likely alter vascular regulation, starving neurons of oxygen when they need it most, Iadecola said. He is now investigating the mechanisms of Aβ and tau action on the vasculature in more detail, asking whether they work in concert or independently.
Barrier Breach
If the brain’s vasculature is damaged in Alzheimer’s, how would doctors know? Zlokovic gave an overview of his ongoing work on just such a biomarker: brain blood flow measured by a technique called dynamic contrast-enhanced MRI. The contrast agent, containing gadolinium, cannot cross the blood-brain barrier unless the barrier is leaky. Using this method, Zlokovic and colleagues previously reported that the barrier starts to become porous with age in cognitively healthy people, and even more so in those with mild cognitive impairment (see Feb 2015 webinar). At the Zilkha conference, Zlokovic told attendees he has preliminary evidence that this permeability at the barrier might have consequences for connectivity of neurons. He is also following up, in people, on a previous mouse study that indicated the ApoE4 genotype makes the barrier more porous (Bell et al., 2012).
“Gadolinium-enhanced imaging of permeability of the blood-brain barrier is a potential biomarker, for both compromise of the barrier and the advent of cognitive impairment and dementia during aging. It is a potentially powerful new tool that I hope is explored further,” said Corriveau.
Iadecola agreed, saying indicators of a leaky blood-brain barrier could be particularly important as an early marker, well before symptoms arise, when the disease process is underway in the background. “That is going to be the future in Alzheimer’s disease,” he said. In addition to the MRIs, Zlokovic noted to Alzforum that he has discovered fluid biomarkers, particularly PDGF receptor β in the cerebrospinal fluid, that can flag barrier breakdown.
Probing Pericytes
Contractile pericytes are crucial to maintaining the blood-brain barrier. If they malfunction in Alzheimer’s, then scientists will need to understand the details of those defects in order to find ways to repair them, Iadecola said. But even before this step, scientists still need to more fully comprehend the normal inner workings of a healthy pericyte. For example, little is known about how a pericyte differs from the endothelial cells that line the brain’s blood vessels. That is where Betsholtz’s work on their basic biology comes in. Betsholtz is profiling pericytes by RNA sequencing of single cells, and analyzing their proteome. Although these cells are very interesting, Betsholtz told Alzforum, researchers still do not understand if they play a major role in AD or are bystanders.
Zlokovic told Alzforum his group also is developing methods to study this question, using a mouse model that expresses the Cre enzyme specifically in pericytes. “This new model will allow us to express or delete any genes specifically from pericytes,” he said. “We can use it to address many questions.”
Pericytes and the rest of the vasculature are likely to get plenty of attention in the future, scientists said. “The neurovascular unit is probably more important than we think,” commented Philip Scheltens of VU University Medical Center in Amsterdam.—Amber Dance



Comments
No Available Comments
Make a Comment
To make a comment you must login or register.