Some 29,000 registrants descended on McCormick Place, the largest convention center in the United States, for the Society for Neuroscience annual meeting, held October 17-21 near downtown Chicago. Among the 18 symposia, 31 minisymposia, 98 nanosymposia, 650 poster sessions, and numerous special lectures was a shrinking but still lively fraction of Alzheimer’s science. Alzforum reporters Madolyn Rogers and Tom Fagan ferreted it out.
Snapshots from the Society for Neuroscience Annual Meeting
At around 29,000 registrants, this year’s Society for Neuroscience annual meeting, held October 17-21 in Chicago, was slightly smaller than those of years past. More noticeably for Alzheimer’s researchers, the number of oral and poster presentations on AD has been shrinking in recent years as well, and this trend continued this year. There were no minisymposia on the SfN subtheme of AD and other dementias, and just seven nanosymposia—down from 11 in 2014. Some scientists lamented that SfN is no longer a must-go for AD researchers. Even so, the nanosymposia, almost 500 posters, special lectures, and satellite events on this research area did yield enough to quench a thirst for the basic science behind neurodegenerative disorders. Below is a snapshot of some of the highlights.
A theme that continued to impress attendees was the role of the immune system in neurodegeneration. In her Presidential Special Lecture, Beth Stevens, a pioneer in this field and recent recipient of a MacArthur fellowship, showed new evidence for how complement proteins of the innate immune system mediate microglial digestion of synapses in developmental disorders such as schizophrenia and neurodegenerative diseases such as AD. This mechanism may underlie Aβ’s ability to damage synapses, since lack of a key complement protein renders the peptide harmless, Stevens said. In other presentations, researchers expanded on the idea that some synapses display an “eat me” signal that invites aberrant pruning in several disorders. Meanwhile, Stevens’ group at Boston Children’s Hospital identified a corresponding “don’t eat me” signal that shields synapses from such gastronomic humiliation.
Bin This.
Silencing Bin1 (right) ramps up endosomes as seen by accumulation of the marker Rab5 (purple), but limits axonal trafficking of BACE1 (green). [Image courtesy of Claudia Almeida, Universidade Nova de Lisboa.]
Microglia and the immune system have risen to prominence in AD research because genome-wide association studies identified risk loci near genes for microglial proteins and immune signaling molecules. In Chicago, scientists reported how they are making inroads into the still-mysterious functional effects of GWAS variants in Bin1, Clu, ABCA7, CR1, and TREM2. Bin1 seems to affect endosomal trafficking (see image), though scientists debated where the protein acts, with one group suggesting it’s in axons and another suggesting synapses. Knockout mice yielded strong evidence that clusterin modulates clearance of Aβ from the parenchyma into blood vessels. An ABCA7 variant appears to alter splicing of the gene, both changing the protein’s structure and reducing its amount. The alternative splice variant also occurs in some people who do not harbor the corresponding GWAS SNP, suggesting that other upstream factors influence production of that splice variant, and that these factors converge to potentially alter microglial phagocytosis of Aβ. Other scientists reported that while alternatively spliced forms of CR1 that correspond to GWAS SNPs are expressed at similar levels in control and AD brains, they bind more tightly to complement factors C1q and C3b. Finally, new evidence that TREM2 tones down tau pathology was in keeping with the idea that microglia mop up unwanted proteins in the brain.
Beyond GWAS hits, scientists have begun to screen panels of wild-type mouse strains to map novel genetic variants that modulate AD-relevant behavioral phenotypes such as learning and memory, or that interact with genes identified in human GWAS. Researchers mapped loci in smarter mice to the gene for a chromatin-binding protein. That regulator seems to controls expression of an autophagy gene that is upregulated in people with AD.
No Clu.
APP/PS1 mice lacking the clusterin gene accumulate vascular A deposits (green, bottom) as opposed to parenchymal plaques found in control mice (top). [Image courtesy of John Fryer, Mayo Clinic.]
Science also continues to learn from mice that model human genetic variation, for example trisomy 21, the cause of Down’s syndrome. At SfN, researchers reported that treating DS model mice soon after birth, or even treating their murine moms prenatally, can normalize brain development and prevent cognitive impairment. If this finding holds up, it would represent a sea change in how researchers view the condition. The data have raised hopes for similar strategies in children with Down’s, though numerous hurdles remain, including finding a treatment safe enough for use during pregnancy. Possible agents for this type of intervention include inhibitors of the kinase DYRK1A, which stimulates inhibitory interneurons, and serotonin selective reuptake inhibitors, such as Prozac, which seemed to normalize brain development when given to pups prenatally or right after birth. Overall, there seemed renewed hope that one day researchers may be able to help brain development in children with the extra chromosome.
Researchers described novel therapeutic approaches for neurodegenerative diseases and updates on therapeutics already in preclinical testing. Immunotherapies for Alzheimer’s, Parkinson’s, and related conditions ran the gamut from antibodies against pyroglutamate-modified Aβ that cleared plaques in AD mouse models and improved their learning and memory, to oligomer-directed antibodies that prevented cross-seeding of tau with α-synuclein, and rescued synapses and movement phenotypes in mouse models of PD. Others reported on small-molecule approaches for AD and PD targeting the protein phosphatase calcineurin, a microglial purine receptor, a metabotropic glutamate receptor, tyrosine kinases, and the Parkinson’s gene DJ-1.
This year’s SfN meeting reflected incremental progress in understanding how trauma affects the brain. Scientists claimed that brain injuries have variable effects depending on factors such as sex, age, and even diet. They proposed underlying mechanisms for cognitive problems associated with traumatic brain injury, such as disrupted autophagy, poor DNA methylation, loss of dopamine signaling, and axonal injury. Other groups identified possible neuroprotective factors, such as a transcription factor that rises after injury to boost expression of antioxidant proteins. Several speakers reported on potential therapeutics, including antioxidant drugs, hyperbaric oxygen therapy, and normalizing calcium flux in neurons, which protected the brain from degeneration when given to animals soon after injury. No consensus emerged; rather, the overall picture was one of a heterogeneous condition that may involve numerous mechanisms and require multiple treatment approaches.
In related events, the RNA Metabolism in Neurological Disease satellite conference, held October 15-16, drew an enthusiastic crowd. This field is buoyed by recent discoveries surrounding C9ORF72 expansions, which lead to expression of aberrant RNAs, and RNA-binding proteins such as FUS and TDP-43 that are linked to motor neuron disease and frontotemporal dementia. Researchers debuted new mouse models of C9ORF72 toxicity. Knockouts had massive spleens and rampant cytokine signaling, indicating a major disturbance to the hematopoietic/immune systems. Mice overexpressing the expanded gene had complex phenotypes, with some struggling with movement while others behaved oddly, evoking frontotemporal dementia. In this respect, the overexpression models appear to reflect the disease spectrum of the C9ORF72 expansions. In a strange twist on the C9ORF72 story, researchers reported that the hexanucleotide-expanded RNAs made from the mutated gene might condense into liquid phase droplets, similarly to proteins prone to aggregate (see upcoming Alzforum Webinar).
At this satellite meeting, researchers also reported on a mouse model of ALS/FTD that harbors a suppressible TDP-43 transgene. When it was turned off after protein deposits had amassed in the brain, the mice cleared the deposits and survived. What’s more, motor neurons that endured the regimen took over from ones that hadn’t, innervating muscle and restoring function. This implies that if TDP aggregates in people could be dissolved, disease may not only be stopped, but patients might even regain some function.
Researchers at the satellite conference were energized by progress toward therapeutics. Gene-silencing technology against C9ORF72 and other proteins involved in neurodegeneration appears to show promise in mice and clinical trials are starting. Attendees were further intrigued by the prospect of turning a natural chaperone into a broad-spectrum disaggregase that attacks several neurodegeneration proteins.
Finally, scientists reported on lesser-known RNA binding proteins that may link to ALS/FTD pathology. RBM45 appears to form nuclear stress bodies distinct from the cytoplasmic RNA stress granules found in ALS, while hnRNPA3 seems to slow production of dipeptide repeats from the C9ORF72 hexanucleotide expansions.
Check back in the coming weeks as Alzforum brings you these stories in depth.—Tom Fagan
Microglia Control Synapse Number in Multiple Disease States
What causes synapses to wither in neurodegenerative diseases such as Alzheimer’s? Some researchers blame reactivation of microglia, which prune unneeded synapses during brain development. At the Society for Neuroscience meeting held in Chicago October 17-21, Beth Stevens of Boston Children’s Hospital, a pioneer in this field, presented new evidence to support the idea that these immune cells of the brain devour synapses in Alzheimer’s and Huntington’s diseases. Other researchers reported that excessive synapse consumption occurs in obesity as well, and that problems with developmental pruning may predispose children to disorders such as schizophrenia and autism. Several talks and posters described the mechanisms by which microglia control the number of synapses, including the identification of a protective “don’t eat me” signal. The topic generated buzz among attendees and appears to be gaining traction among neuroscientists, as evidenced by multiple groups now reporting similar data.
“This research has changed our understanding of microglia and their potential roles in neurodegenerative and neurodevelopmental disorders … the field is moving forward very fast and the new developments are exciting,” Jonathan Kipnis of the University of Virginia, Charlottesville, wrote to Alzforum. He studies interactions between immune cells and neurons. Ben Barres of Stanford University, California, agreed, noting, “The work not only provides a detailed mechanistic pathway for understanding how synapses are lost, but also one that is therapeutically targetable.”
Immune Molecules Dapple Brain.
Three different MHCI molecules (red, blue, and green) reveal distinct expression patterns in the brains of 6-week old mice. [Image courtesy of Carla Shatz and Science/AAAS.]
The idea that the immune system regulates synapse number dates back only about a decade. At that time, Carla Shatz of Stanford reported excessive synaptic plasticity in mice that lacked the major histocompatibility class I (MHCI) protein or its receptor PirB. Meanwhile Stevens, then working with Barres, described a synaptic bounty in young mice lacking the C1q or C3 proteins of the immune complement system, which signal microglia to snip synapses (see Nov 2007 conference news). Other groups have since confirmed that microglia touch, regulate, and engulf synapses (see Nov 2010 news; Dec 2013 news). Astrocytes get into the act as well, chewing up synapses during development using a different suite of receptors (see Dec 2013 conference news).
Some studies hinted that synapse ingestion might ramp back up in Alzheimer’s disease. Shatz reported synapse retention in AD model mice lacking PirB (see Sep 2013 news). Likewise, Cynthia Lemere at Brigham and Women’s Hospital, Boston, in collaboration with Stevens, found more synapses and better memory in APPPS1 mice lacking C3 (see Aug 2013 conference news). Recently, Stevens’ group tied elevated C1q to synaptic disconnection in the J20 mouse model (see Mar 2015 conference news).
At SfN, Stevens expanded on these data. In a Presidential Special Lecture before a packed hall, she noted that synapses in AD model mice bristle with excess C1q even at young ages, before amyloid plaques form. The fact that C3 knockouts are protected from the synaptic carnage normally seen in AD mice suggests that complement proteins mediate elimination, she said. To find out if Aβ plays a role, the researchers injected oligomers of the peptide into the ventricles of wild-type mice. Within three days, they saw a dramatic rise in C1q at synapses in stained brain sections from hippocampus, as well as a steep drop in synapse number. Microglia likely gobbled up these structures, as the researchers saw fluorescently labeled synapses inside these immune cells after Aβ treatment. Again, complement appeared to mediate loss, as mice missing the C1qA subunit maintained their synapses just fine after Aβ injections. Likewise, blocking C1q by passive immunotherapy before injecting Aβ protected synapses from destruction in live mice. Stevens did not say if the immunotherapy alone had any effect. In hippocampal slices, pre-treatment with the antibody preserved long-term potentiation. Altogether, the data implicate microglia and complement as the culprits in the disappearance of synapses in AD models, Stevens said. She added that it is still unknown what causes C1q to rise in these models, or how Aβ and C1q work together.
What about other neurodegenerative diseases? In a separate talk, Daniel Wilton in Stevens’ group extended the findings to mouse models of Huntington’s disease. In this condition, caused by a polyglutamine expansion in the huntingtin protein, people develop both movement and mental problems and striatal degeneration with age. Wilton used the Q175 HD mouse, which loses synapses but not neurons, and probably models early disease. By three months of age, C1q accumulated at synapses, and microglial activation ran wild in the striatum and motor cortex, he found. Microglia in these mice contained more synaptic material than did those in control animals, suggesting they were gorging on these connections. Crossing Q175 mice with CR3 knockouts protected synapses, as did injecting a C1q antibody every other day for a month. In future work, Wilton will examine whether protecting synapses slows the progression of the disease and helps preserve motor functions. When asked if the same mechanism occurs in human disease, Wilton said preliminary data reveal more complement at synapses in postmortem brain slices from HD patients than from healthy controls. He is also measuring complement levels in cerebrospinal fluid samples from patients to see if these proteins spike before synapses start to disappear.
Microglia may even thin out synapses in other health conditions. Obesity, a risk factor for cognitive decline and dementia, boosts inflammation throughout the body (see Sep 2015 news; AlzRisk analysis). Alexis Stranahan of the Medical College of Georgia, Augusta, wondered if that inflammation might activate microglia. To test this, she compared mice that ate chow containing 60 percent fat to those on a 10 percent fat diet for 12 weeks. The tubby mice had more C1q at synapses, and more microglia crowded those synapses, she found. In culture, microglia from the obese mice were more mobile and devoured more synapses, suggesting they had been activated for phagocytosis (see Hao et al., 2015). Inhibiting the C3 complement receptor prevented the synaptic stripping, Stranahan said. C3 is activated by C1q.
Intimate Contact.
Microglial processes (green) touch dendritic spines (yellow) in cultured hippocampal slices. Scale bar, 1 μm. [Image courtesy of Thomas Pfeiffer and Valentin Naegerl, University of Bordeaux.]
In addition to these late-life disorders, perturbations in synapse maintenance may also contribute to neurodevelopmental conditions such as schizophrenia and autism—though whether the problem is too few synapses or too many remains unclear. At SfN, Cornelius Gross of the European Molecular Biology Laboratory, Monterotondo, Italy, made a case for the latter. He reported that mice lacking the microglial fractalkine receptor, CX3CR1, maintained a greater number of feeble synapses. In these mice, brain activity coordinated poorly. The animals repeated actions over and over, and interacted poorly with littermates, traits reminiscent of autism (see Zhan et al., 2014). Why might this be? Fractalkine is a neuronal transmembrane protein that attracts microglia and is highly expressed during synaptogenesis. The CX3CR1 knockouts have fewer microglia in their brains, which may lead to deficient synaptic pruning, Gross said. Fractalkine expression in apoptotic neurons has previously been found to stimulate microglial phagocytosis (see Sokolowski et al., 2014). Gross noted that the failure to eliminate some synapses seems to prevent the strengthening of others, as though there is some limiting resource. This leads to overall weak synaptic connectivity, a pattern also seen in autism and other neurodevelopmental disorders. Together, the results hint that insufficient synaptic pruning by microglia could contribute to neurodevelopmental problems.
Other evidence implicates excessive loss of synapses. Stevens described a genetic study of 34,000 people with schizophrenia by Steve McCarroll of Harvard Medical School. It correlated polymorphisms near the C4 complement gene with disease risk. In particular, McCarroll saw a connection with the allele for the C4A subunit. This gene has undergone extensive duplication and deletion through the years, such that the number of copies vary widely from one person to another. He found that people lacking the allele for the C4A subunit had a low risk of the disease, whereas those with multiple copies had high risk. C4 participates in synaptic pruning just as C1q and C3 do, Stevens noted. The data suggest overzealous synapse elimination might underlie schizophrenia.
Similarly, Kim McAllister at UC Davis pointed a finger at excessive loss of synapses in neurodevelopmental disorders. Myka Estes in McAllister’s lab studies a mouse model of maternal immune activation (MIA), in which researchers inject a viral mimic into pregnant females to stimulate inflammation (see Malkova et al., 2012). Some researchers believe maternal inflammation sets the stage for later schizophrenia in children, and indeed, the adult offspring of these mice exhibit behaviors reminiscent of autism and schizophrenia. In 2013, McAllister and colleagues found that the offspring also had more major histocompatibility class I (MHCI) marking synapses, and only half as many synapses as controls, McAllister reported (see Elmer et al., 2013).
In Chicago, McAllister said that the researchers had linked these changes to a doubling of the cytokine IL-1β in the offspring. In support of a role for IL-1β, treating cultured neurons with this cytokine boosted MHCI and reduced the number of synapses on the cell surface. Conversely, lowering MHCI preserved synapses from the harmful effects of IL-1β. McAllister hypothesized that maternal infection pumps up IL-1β levels, leading to elevated MHCI and a corresponding loss of synapses. IL-1β is also elevated in the blood and brain of people with schizophrenia and autism, she noted.
Data from other groups supports a role for IL-1β in synaptic pruning. In a poster, Nasr Farooqi from the lab of Edward Ruthazer at McGill University reported that the cytokine prevented synapse stabilization in zebrafish larva. Stimulating inflammation in these animals caused microglia to activate and pump out IL-1β. Axons initially became more mobile and put out more feelers, but in succeeding days their axonal arbors thinned out, suggesting that new synapses failed to stabilize, said Ruthazer. Knockdown of IL-1β or deletion of microglia prevented this loss. Intriguingly, IL-1β has also been implicated in loss of synapses in obese mice (see Erion et al., 2014).
Can synapses protect themselves from overeager microglial pruning? It appears so. Emily Lehrman in Stevens’ group described a protective signal. In the periphery, transmembrane CD47 acts as a “don’t-eat-me” signal for many cell types, binding to signal regulatory protein α (SIRPα) on macrophages to inhibit engulfment. Lehrman wondered if CD47 could play a similar role in the brain. Consistent with this, she found high CD47 at some but not all synapses in the visual system of mice five days after birth, during the peak of synaptic pruning, as well as high SIRPα in microglia. By two months of age, both CD47 knockouts and SIRPα knockouts had lost more synapses than wild-type mice, fitting with the idea that these are protective molecules, she reported. In addition, blocking SIRPα boosted microglial appetites for synapses in vitro. In future work, Lehrman will assess whether these changes in synapse number have consequences for behavior. Other studies have found that CD47 knockouts are less sociable, she noted (see Koshimizu et al., 2014).
Overall, the emerging picture suggests that microglia affect synapses in both development and disease states. Could they regulate synaptic plasticity in healthy adults as well? Thomas Pfeiffer, working in Valentin Nägerl’s team at the University of Bordeaux, France, presented some mouse evidence that they might. Using two-photon imaging in hippocampal slices from healthy adults, he found that after stimulating long-term potentiation of synapses, microglia scanned their environment more actively, putting out more processes. Surprisingly, the microglia made fewer contacts with synapses, but maintained each contact for longer. Pfeiffer could prevent these microglial changes by blocking NMDA receptors, which mediate synaptic remodeling, demonstrating that the immune cells were responding to LTP. Next, he will use super-resolution microscopy to investigate whether synapses touched by microglia share some common feature, and what happens to them after this contact, he told Alzforum.
Researchers agree that they have only begun to scratch the surface of how microglia and astrocytes may affect synapses. “The field is in its infancy,” Kipnis wrote to Alzforum. “It may be too early to understand exactly what we are seeing, but it’s something we were previously blind to.”—Madolyn Bowman Rogers
Might Normalizing Brain Development Help in Down’s Syndrome?
Can the cognitive impairments of Down’s syndrome be prevented? A decade ago, no one even asked this question. Cognitive difficulties were assumed to be an inevitable consequence of carrying an extra copy of chromosome 21. With the advent of mouse models, however, researchers have begun to dissect what goes wrong in the brain during development and address how to correct it. At a symposium at the Society for Neuroscience annual meeting held in Chicago October 17-21, speakers made the case that early postnatal or even prenatal treatment can restore normal brain structure and size and prevent cognitive defects in mice that model the disorder. With additional research, some of these approaches might be ready for testing in human clinical trials within three to five years, co-chairperson Diana Bianchi of Tufts Medical Center, Boston, wrote to Alzforum. Chairperson Renata Bartesaghi of the University of Bologna, Italy, emphasized that for the first time, researchers hold hope that they may be able to help people with Down’s syndrome to live more independently.
“This is a very exciting time in Down’s syndrome research,” said Ted Brown, who heads the Medical Genetics Laboratory at the Institute for Basic Research, Albany, New York. Michael Harpold of the LuMind Foundation, a non-profit in Marlborough, Massachusetts, that supports Down’s syndrome research, said he was encouraged by the progress in this area. He noted that several drugs targeting cognition are already in clinical trials for children and adults with Down’s syndrome (for review, see Gardiner, 2014). Prenatal treatments, however, would represent a sea change in how researchers approach the condition.
Restored Neurogenesis. Adult mice that model Down’s syndrome (middle) have fewer granule cells (purple) compared to controls (left); prenatal treatment with fluoxetine (right) restores cell number. [Courtesy of Renata Bartesaghi.]
Down’s syndrome is fairly common, occurring in about one in 700 live births. People who are affected carry one extra copy each of about 360 genes found on chromosome 21, disrupting numerous processes including the cardiac, digestive, and motor systems. The most notable feature of Down’s, however, is cognitive impairment. The brain develops abnormally in utero. Fewer neurons are born, and they segregate to the wrong cortical layers. By 15 weeks of pregnancy, brains of DS children are significantly smaller than those of other babies, with a shrunken cortex and cerebellar volume about 80 percent of normal.
The first useful mouse model of Down’s syndrome, Ts65Dn, was developed in 1993 (see Davisson et al., 1993). It carries extra copies of many of the genes on human chromosome 21, though it is not a perfect genetic match for the human condition. The mice share many physical and behavioral features of DS, and mirror the defects in early brain development (see Feb 2000 news; Tyler and Haydar, 2013). Speakers agreed that this model, and others that followed, revolutionized DS research. “Ts65Dn broke open the door,” said Tarik Haydar from Boston University.
Surprisingly, research in this mouse suggested that its complex learning and memory defects could be corrected if relatively simple treatments, such as neurotrophic factors or activators of sonic hedgehog, a crucial developmental signaling molecule, were given early enough (see Roper et al., 2006; Toso et al., 2008). At SfN, Bartesaghi presented another such approach. She noted that the serotonin signaling system is suppressed in DS brains from the embryonic stage, and wondered if selective serotonin reuptake inhibitors might normalize it. She treated newborn Ts65Dn mice with 5-10 mg/kg fluoxetine (Prozac) for two weeks, which is roughly equivalent to doses people take for depression. Treated mice mustered as many neurons and synapses as control mice, and had normal thickness of the granule cell layer of the cerebellum (see image above). At six weeks old they learned as well as wild-type mice in fear conditioning tests, Bartesaghi reported. In addition, neurons had wild-type levels of the β-CTF fragment of APP, and showed no signs of the swollen endosomes that accumulate in AD brains and may be a precursor to AD-like degeneration (see Bianchi et al., 2010; Stagni et al., 2015; May 2011 news). The data suggest that the early treatment not only normalized serotonin transmission, but neurodevelopment and APP processing as well.
Next, Bartesaghi and colleagues treated pregnant Ts65Dn mice with 10 mg/kg fluoxetine, starting 10 days after conception and ending at birth. The authors did not measure fluoxetine levels in the pups’ brains, however, as with postnatal treatment, the offspring had normal cerebellar thickness and neuron numbers at birth and six weeks of age, based on counts from numerous brain regions. They also behaved like wild-type mice at six weeks old, performing normally in fear conditioning tests and displaying none of the hyperactivity that characterizes the Ts65Dn mice (see Guidi et al., 2014).
The data were well-received, with one scientist saying he was “blown away” by the findings. However, the results are still preliminary. Bartesaghi noted she has not yet conducted a thorough safety study, including assaying for the cardiac problems that can occur with SSRI use, nor has she looked for a dose response or tested the drug in other mouse models. In answer to an audience question, Bartesaghi said it is not yet clear whether there is a difference between treating prenatally or early postnatally, although she suggested that prenatal treatment is likely to have a more widespread effect on the brain.
Could prenatal treatments from mice be applied to people? Doctors would first have to diagnose DS early enough. Bianchi pointed out that new cell-free DNA screening tests, available since 2011, can detect DS at 11 weeks gestation with more than 99 percent accuracy. Though results still need to be confirmed with invasive testing, this could provide a window for early intervention, she noted. However, drugs for prenatal treatment would need to have an excellent safety profile, and be able to cross the placenta as well as the blood-brain barrier.
Bianchi outlined an approach to finding new drug candidates. She analyzed gene expression using RNA from the amniotic fluid of DS babies, and saw from multiple samples a consistent pattern in 15- to 16-week-old fetuses. Comparison of the gene-expression pattern to that of normal babies revealed that oxidative stress pathways were particularly disrupted (see Slonim et al., 2009). This matches data from studies of Ts1Cje mice that model the disease (see Guedj et al., 2015). Bianchi then looked for drugs that would normalize expression. She used the Connectivity Map created by the Broad Institute of MIT and Harvard, which profiles 1,309 FDA-approved drugs by microarray analysis to show how each one affects gene expression. From this list, Bianchi and colleagues identified 47 compounds that targeted the major pathways, including oxidative stress, ion transport, and G protein signaling, which were disrupted in both people with DS and Ts1Cje mice. The researchers then used analytical software to rank these drugs by their expected ability to correct gene expression.
One of the top candidates was apigenin, an antioxidant found in leafy green vegetables, citrus fruits, and chamomile tea. Apigenin is a dietary supplement under investigation as an anti-cancer agent. It purportedly promotes neurogenesis (see Taupin, 2009). Bianchi and colleagues tested the compound on cultured DS cells and found it prevented the “unspooling” of DNA. This is when the nucleic acid unwinds from its dense chromatin packaging—a known marker of oxidative stress. The researchers then fed 200 mg/kg apigenin daily to pregnant Ts1Cje mice. During the first few weeks after birth, the offspring remembered better and reached developmental milestones sooner than those born of untreated controls. Apigenin did not improve all DS symptoms, however, as motor skills remained poor. In future work, the researchers will test apigenin in other mouse models, and will also examine whether combination drug treatment produces better results, Bianchi said.
Other researchers are taking a different approach to normalizing brain development. Previously, researchers identified excessive synaptic inhibition as one of the primary problems in mouse models of DS (see Siarey et al., 1997; Kleschevnikov et al., 2004; Mar 2015 news). At SfN, Jean-Maurice Delabar of Paris Diderot University, France, noted that the kinase DYRK1A helps control the balance between excitation and inhibition in the brain (see Souchet et al., 2014). People with Down’s syndrome, as well as Ts65Dn mice, carry an extra copy of this gene. Moreover, transgenic mice that overexpress DYRK1A generate excessive inhibitory interneurons and have disorganized brain layers, fewer neurons, and connectivity problems (see Najas et al., 2015). The transgenics demonstrate that this single gene can recapitulate many of the problems in DS brains. The kinase appears to affect other aspects of Down’s syndrome as well, such as skeletal abnormalities (see, e.g., Blazek et al., 2015). The data suggest the kinase might be a promising target for restoring healthy brain function in Down’s syndrome, Delabar said.
Delabar tested this approach by treating adult Ts65Dn mice for one month with epigallocatechin gallate (EGCG), a polyphenol found in green tea. EGCG inhibits DYRK1A (see Bain et al., 2003; Guedj et al., 2009). Hippocampal slices from treated animals had better LTP and the mice had sharper memories, though they still sported more inhibitory interneurons than usual, Delabar reported. He is now testing EGCG in pregnant Ts65Dn mice. By contrast with postnatal treatment, preliminary results suggest that prenatal treatment lowered the number of inhibitory interneurons in the offspring to normal levels, he said. In a pilot clinical study, adults with Down’s syndrome who took EGCG scored better on memory tests (see De La Torre et al., 2014).
Mara Dierssen of the Center for Genomic Regulation, Barcelona, who collaborated with Delabar on several studies, made a case for combining EGCG treatment with cognitive stimulation. When mice are housed in cages with toys, they perform better on learning and memory tests. Dierssen previously found that such environmental enrichment can normalize DYRK1A activity in mice overexpressing the gene, and hypothesized that it might have synergistic effects with EGCG (see Pons-Espinal et al., 2013). She found that the two treatments together were more effective than either alone at boosting memory in six- to seven-month-old Ts65Dn mice. The combination treatment also preserved cholinergic neurons, but the single interventions did not.
In children with DS, computerized cognitive training has been used to improve memory (see Bennett et al., 2013). Dierssen tested a combination of pharmacological and cognitive treatments in a Phase 2 clinical trial of 31 teens and young adults with Down’s syndrome. Participants took either EGCG supplements or placebo for three months, and some in each group also took part in computerized cognitive training. As with the mice, those who received both interventions demonstrated more improvement on memory tests than those who had only one, Dierssen reported. Moreover, functional MRI revealed that the regions of their brains worked together more smoothly, suggesting better connectivity after treatment. Improvements lasted at least three months after treatment stopped. DYRK1A may prime cells so that the effects of environmental enrichment are more stable, Dierssen suggested.
Dierssen noted that DYRK1A also affects amyloidogenesis, and so could be targeted in adults with DS to try to slow amyloid-β accumulation (see Coutadeur et al., 2015; Naert et al., 2015). Because people with DS carry three copies of APP, they develop amyloid pathology by their 40s (see Jun 2011 news). Intriguingly, EGCG has been found to block amyloid formation and has been tested, but never developed, as an Alzheimer’s treatment (see Oct 2005 conference news; May 2008 news).
Carmen Martinez-Cué of the University of Cantabria, Santander, Spain, focused on slowing AD pathology in her talk. She is trying several strategies in DS models. In one approach, she treated adult Ts65Dn mice with melatonin for seven months. Melatonin regulates circadian rhythms, but also suppresses inflammation, oxidative stress, and APP while boosting neurogenesis, suggesting it might help protect aging brains. Treatment lowered lipid peroxidation and protein damage in hippocampus, Martínez-Cué reported. In addition, treated animals had better cognition along with improved neurogenesis and LTP in the hippocampus, though not to the level of that in normal controls (see Corrales et al., 2013; Corrales et al., 2014). In a second strategy, Martinez-Cué developed a neutralizing antibody to the proinflammatory cytokine IL-17. She administered this antibody to Ts65Dn mice from five to 12 of months of age to try to quiet neuroinflammation. Treated mice displayed more neurogenesis and less Aβ. They performed better in a water maze than untreated littermates, but again not to control levels.
In the end, many different approaches may be needed to combat all the symptoms of Down’s syndrome, Bianchi told the crowd. These problems span the gamut from early brain abnormalities to muscle weakness to neurodegeneration, and may each present different windows for intervention. Randall Roper of Indiana University-Purdue University, Indianapolis, wrote to Alzforum, “The big hurdle to me is to show how and where potential therapies are working at the cellular and molecular level … It is important to have sufficient funding to continue necessary basic science research before large-scale clinical trials begin.” Research into these areas is accelerating. Bianchi noted that scientists founded the non-profit Trisomy 21 Research Society in 2014, and this group held its first meeting June 4-7 in Paris. In advance of the SfN symposium, the presenters wrote up a preview of their talks, which appeared in the October 14 Journal of Neuroscience.—Madolyn Bowman Rogers
Current treatments for Parkinson’s patients help restore movement, but do not slow the underlying brain degeneration. Researchers are pursuing strategies to curb the accumulation of α-synuclein in hopes that this may delay disease progression. At the Society for Neuroscience annual meeting in Chicago October 17-21, scientists reported that two different drugs that promote α-synuclein clearance have completed Phase 1 safety testing, with biomarker results suggesting they are hitting their targets. Both are headed to Phase 2. Others reported on immunotherapy approaches in preclinical testing, including a strategy to disrupt α-synuclein deposits by targeting, of all things, tau oligomers. Meanwhile, after a decade-long hiatus, cell replacement strategies are gathering steam once again. Data on stem cell approaches, including one poised to enter Phase 1, intrigued the SfN crowd. Whether any of these treatments will pan out remains to be seen, but the talks reflected new momentum, with numerous disease-modifying therapies having entered the pipeline for Parkinson’s.
“I’m a huge fan of tackling α-synuclein. I think it’s the No. 1 target for Parkinson’s therapeutics,” said Jeffrey Kordower, Rush University, Chicago, who chaired a Parkinson’s symposium. Kordower noted that lowering α-synuclein might improve not only movement, but also non-motor symptoms, such as cognitive decline, for which no treatments currently exist. The non–motor symptoms typically lead family members to admit someone with Parkinson’s to a nursing home, he added.
Target Tau to Treat PD? In untreated PD mice (left), and in those treated with an antibody to all forms of tau (middle), reactive astrocytes (red) swarm tau deposits (green). Mice treated with a tau oligomer-specific antibody (right) do not activate astrocytes. [Courtesy of Julia Gerson.]
In both Parkinson’s and dementia with Lewy bodies, deposits of aggregated α-synuclein accumulate in the brain. Because these aggregates, either as smaller oligomers or larger Lewy bodies, are believed to underlie disease symptoms, many groups are seeking ways to bust them up. At SfN, Fernando Pagan of Georgetown University, Washington, D.C., claimed the cancer drug nilotinib may do just that. Nilotinib, FDA-approved to treat adult chronic myeloid leukemia, inhibits the tyrosine kinase Abl. Abl phosphorylates α-synuclein and protects it from degradation via autophagy (see Mahul-Mellier et al., 2014). Inhibiting Abl seems to stimulate autophagic clearance of α-synuclein, Pagan said. His collaborator Charbel Moussa at Georgetown tested nilotinib in A53T mice, which overexpress mutant human α-synuclein and develop Lewy bodies. In these animals, 10 mg/kg of nilotinib given every other day for six weeks promoted autophagy of α-synuclein and of phosphorylated tau, lowering levels of these proteins by about half (see Hebron et al., 2013; Hebron et al., 2013; Hebron et al., 2015). Nilotinib treatment also pumped up brain dopamine levels and improved motor skills and cognition, Pagan reported at SfN.
Based on these data, Pagan and colleagues launched a Phase 1 study of the drug in 12 people with advanced PD or DLB at Georgetown University Hospital. For six months, participants received either 150 or 300 mg nilotinib daily, or about one-quarter the dose used for cancer treatment. At SfN, Pagan reported that participants tolerated the drug well, with no serious side effects. Since nilotinib can cause irregular heart rhythms, people with abnormal cardiac rhythms were excluded from the trial. At doses used in cancer treatment, nilotinib can also suppress the production of blood cells in the bone marrow. The participants maintained normal heart rhythms and blood cell production, Pagan reported.
The researchers tracked a suite of biomarkers. They found that plasma levels of α-synuclein dropped by more than half. CSF levels, which normally fall as Parkinson’s progresses, did not change. Tau and phosphorylated tau in CSF, which typically rise as cognition worsens, dropped. Other proposed CSF markers of neurodegeneration, such as S100B, also fell. Meanwhile, CSF dopamine levels climbed, causing the researchers to lower the dose of L-dopa given to some patients. Nilotinib appeared in CSF as well, indicating the drug entered the brain.
Participants self-reported that they functioned better on nilotinib, though that should be interpreted cautiously because there was no placebo control, the researchers said. Ten of the 11 participants who completed the trial reported meaningful improvements in their ability to speak, walk, and conduct daily activities, Pagan said. Those with DLB, or in earlier stages of PD, gained the most benefit. Improvements disappeared as soon as the drug was withdrawn, he added. The researchers plan to take the drug into Phase 2 in 2016.
Meanwhile, Curt Freed of the University of Colorado, Denver, presented a different approach to clearing α-synuclein. The Parkinson’s gene PARK7 encodes the protein DJ-1, whose many functions include protecting cells against oxidative stress and eliminating misfolded proteins. In primary neuronal cell cultures from A53T mice, DJ-1 blocks aggregation of mutant α-synuclein and prevents its toxicity (see, e.g., Zhou and Freed, 2005; Zondler et al., 2014). Looking for molecules that would increase DJ-1 expression, Freed and colleagues found that the histone deactetylase inhibitor phenylbutyrate roughly tripled DJ-1 levels. The FDA approved phenylbutyrate to treat rare metabolic disorders that prevent children from synthesizing urea. In mouse models of PD and DLB, the drug protected dopaminergic neurons and preserved motor and cognitive functions (see Zhou et al., 2011; Roy et al., 2012). Phenylbutyrate seemed to boost clearance of α-synuclein from the brain, since brain concentrations fell while plasma levels of the protein nearly doubled in treated mice, Freed noted.
Freed led a Phase 1 three-week biomarker study of the drug in 20 PD patients and 20 controls. Participants took 20 g per day, in the form of one teaspoonful of a liquid formulation with each meal. As in mice, treatment nearly doubled plasma α-synuclein, suggesting the drug promoted clearance from the brain. No safety issues cropped up, Freed said. This trial did not assess efficacy. The scientists are planning a Phase 2 trial to start in 2016, he wrote to Alzforum.
Phenylbutyrate has also been investigated for other neurodegenerative disorders, though it does not appear to be in any current trials for these indications. It was reported safe in a 2009 Phase 1 ALS trial (see Cudkowicz et al., 2009), and gave some hints of biomarker efficacy in a Phase 2 Huntington’s trial (see Oct 2011 news). In addition, phenylbutyrate treatment was reported to rescue cognition in Tg2576 AD mice (see Dec 2008 conference news). Freed noted that until recently, the drug was available only in pill form as sodium phenylbutyrate, so to reach the therapeutic dosage of 10 to 20 grams, participants would have to swallow 20 to 40 large pills each day. This may have curtailed interest in pursuing the therapy, he speculated.
Other research groups are trying to harness the immune system to sweep up α-synuclein aggregates. Antibodies against the protein restrict cell-to-cell transmission of α-synuclein in mice, suggesting immunotherapy might slow progression of PD (see Jun 2014 news). At least two anti-α-synuclein immunotherapies are in clinical trials, and more companies are developing such treatments (see Mar 2015 conference news and PRX002).
At SfN, Julia Gerson, who works in the lab of Rakez Kayed at the University of Texas Medical Branch, Galveston, described a different approach to Parkinson’s immunotherapy. Instead of targeting α-synuclein, she went after tau oligomers. Tau and α-synuclein oligomers occur together in Lewy bodies, and have been shown to cross-seed (see Apr 2003 news; Jul 2013 news; Lasagna-Reeves et al., 2010). The researchers previously generated a tau oligomer-specific antibody (TOMA) that crosses the blood-brain barrier and protects mouse models of tauopathy (see Castillo-Carranza et al., 2014; Castillo-Carranza et al., 2014).
Gerson injected a single dose of 120 μg of TOMA into the bloodstream of 17 seven-month-old A53T mice. This is the age when the mice start developing Lewy bodies and muscle weakness, which quickly progresses to paralysis. The authors waited two weeks, then tested behavior. The treatment preserved memory in the object recognition test as well as maintained gait. Treated mice built nests as ably as wild-types did, whereas untreated controls did not. In the brain, treated mice had fewer tau oligomers, more dopamine and dopaminergic neurons, and more synaptic proteins than untreated A53Ts, Gerson reported. The benefits seemed specific to this antibody, because treatment with an antibody that recognizes all forms of tau exacerbated behavioral defects, while a control IgG had no benefits, she added (see image above). Gerson is aging treated mice for 12 months to examine the long-term effects of treatment on behavior and brain pathology. Eventually, she would like to test a combination immunotherapy against both tau and α-synuclein, she said.
While many of the Parkinson’s immunotherapies under investigation employ antibodies, Marika Doucet of the University of Oxford, U.K., chose to pursue an active immunization strategy. Working in Richard Wade-Martins’ group, Doucet expressed fragments of α-synuclein on the surface of a virus-like particle (VLP) derived from a bacteriophage. Presenting the antigens in this way stimulates the immune system to generate antibodies against them. She tested the immunogenicity of three different α-synuclein fragments, one each from the C-terminus, middle, and N-terminus of the protein. Every month for two to three months, Doucet subcutaneously injected 20 μg of each VLP construct into SNCA-OVX model mice. These animals express wild-type human α-synuclein (see Janezic et al., 2013).
The vaccines based on the middle and C-terminal portions of α-synuclein both stimulated production of antibodies that recognized Lewy bodies in postmortem tissue from PD brains, she reported. After two to four months, titers were high enough that the antibodies would, in theory, cross the blood-brain barrier, although the researchers did not directly measure antibody levels in brain. Mice appeared healthy, with normal spleen, blood count, and body weight. However, the researchers saw no change in brain levels of aggregated or oligomeric α-synuclein. Doucet believes the vaccination protocol might have been too short and is now testing a one-year protocol.
Instead of clearing α-synuclein, other researchers are focusing on replacing dopaminergic cells damaged by the disease. This approach is unlikely to slow progression or improve non-motor symptoms, but it can lower the need for L-dopa treatment. Cell replacement fell out of favor a decade ago due to the inconsistent results and side effects seen with fetal dopaminergic grafts, but it has been buoyed by recent reports that some of these grafts remained healthy and provided motor benefits for as long as 20 years (see Jan 2014 news; Jun 2014 news). Now, many groups are turning to stem cells as a more controllable source for transplanted cells.
At SfN, Rodolfo Gonzalez of International Stem Cell Corporation, Carlsbad, California, described an approach using stem cells derived from unfertilized human oocytes by way of parthogenesis (see Jan 2002 news). Led by Eugene Redmond and Ruslan Semechkin, Gonzalez stimulated oocytes to divide to the blastocyst stage, then extracted pluripotent stem cells. These cells have the advantage of possessing two identical copies of every gene, including the human leukocyte antigen (HLA) gene, making genetic matching with patients easier, Gonzalez noted. The researchers had previously developed a protocol to differentiate these pluripotent cells into neural stem cells (see Gonzalez et al., 2013). The FDA has approved these cells for clinical trials, Gonzalez said. In a previous proof-of-concept study in 16 rats and two monkeys whose dopaminergic neurons were killed with toxins, the neural stem cells successfully engrafted into brain and pumped out dopamine (see Gonzalez et al., 2015).
At SfN, Gonzalez presented new data from a yearlong study in 20 African green monkeys. The researchers first injected the neurotoxin MPTP intramuscularly for one month to deplete dopaminergic neurons, then injected either 10 million neural stem cells, 20 million, or vehicle control into the striatum. All the monkeys that received stem cells had more surviving dopamine neurons in the substantia nigra at the end of the study than the control animals did, Gonzalez reported. However, only the low-dose group had more dopaminergic axons, higher dopamine levels in the striatum, and improved motor skills compared to untreated animals. None of the monkeys developed dyskinesia. None developed tumors, a potential side effect of stem cell implantation. The company plans to start a Phase 1/2a clinical trial in late 2015 in Australia and the United States., Gonzalez said. The trial will enroll patients in the early stages of PD.
Clinical trials of these cells might be premature, Kordower told Alzforum. He believes more basic research should first determine what type of cells these NSCs become, how many of them survive, and what exactly they do in the brain. Gonzalez did not address these issues in his talk, and could not be reached for comment. Kordower also works on stem cell programs for Parkinson’s.
Some groups in the stem cell field are transplanting mesenchymal stem cells (MSCs) into the brain rather than embryonic or pluripotent ones. Adult mesenchymal stem cells come from bone marrow and other tissues, making autologous transplant possible, and can be coaxed to differentiate into many cell types. In Chicago, Ryan Welchko in Gary Dunbar’s lab at Central Michigan University, Mount Pleasant, described one such approach. By expressing three transcription factors—Ascl1, Lmx1a, and Nurr1—in MSCs, the researchers induced them to differentiate into cells that resemble dopaminergic neurons. The cells expressed tyrosine hydroxylase, a key enzyme for producing dopamine, as well as dopamine transporters, and pumped out dopamine in culture.
To test the cells, Welchko and colleagues injected the toxin 6-OHDA into the striatum of one brain hemisphere of rats to kill dopaminergic cells. These animals developed poor limb control and a shuffling, slow gait reminiscent of Parkinson’s patients. After eight weeks, the researchers injected the differentiated cells into the brain. Two months later, they saw improvements in movement. Treated rats used their forelimbs for balance as well as wild-types, Welchko said. Typically, animals lesioned in one brain hemisphere spin in circles after being given the dopamine agonist amphetamine because only one side of their brain responds to the drug, but the treated animals spun less, indicating some restored dopaminergic function on the lesioned side. Next, Welchko will examine the brains of these animals to find out if injected cells survived and integrated into the brain circuitry.
With all of these strategies, potential pitfalls lurk as well. Kordower pointed out that because α-synuclein regulates synaptic function, lowering it too far could harm cognition. For cell-based therapies, researchers need to figure out how to avoid the dyskinesias that plague dopamine replacement approaches. Though the research still has a long way to go, speakers agree that scope of current therapeutic strategies has energized the field and generated a renewed sense of optimism for treating the pathology underlying Parkinson’s.—Madolyn Bowman Rogers
Alzheimer’s Risk Genes Give Up Some Secrets at SfN
Large genome-wide association studies have brought the number of Alzheimer’s disease risk genes to more than 20, but how do those common genetic variants modify risk? That the basic function of many of these genes remains shrouded in mystery has not helped scientists answer this question, but at this year’s Society for Neuroscience meeting, held October 17-21 in Chicago, a little more of the veil started to slip. Much as researchers initially expected that these 20 genes would throw the field open wide beyond the amyloid hypothesis, thus far, it appears that some GWAS genes help regulate the two hallmarks of AD: plaques and tangles.
Many labs have used knockout approaches to study GWAS hits, a caveat since most of the common polymorphisms that associate with AD weakly modulate gene transcription and alter risk by as little as 10 percent. Nevertheless, data from knockouts may be informative. As Steve Estus, University of Kentucky, Lexington, pointed out, just because GWAS SNPs have weak effects doesn’t mean we shouldn't target those genes for therapeutic purposes. “If we can pharmacologically modulate those genes by, say, 80 percent, then we may achieve a significant clinical effect,” he said.
Case in point, the gene for clusterin (aka Apolipoprotein J). Researchers in John Fryer’s lab at the Mayo Clinic, Jacksonville, Florida, crossed Clu knockouts with APP/PS1 mice. Graduate student Aleksandra Wojtas reported that the homozygote knockouts shifted Aβ deposition from the parenchyma to the blood vessels, where it deposited to cause cerebral amyloid angiopathy. Other researchers were struck by the stark contrast between knockouts and controls, some calling it the cleanest data they’d seen at the meeting (see image below).
Cluless.
Without clusterin, APP/PS1 mice accumulate Aβ predominantly in the blood vessels (bottom). [Image courtesy of John Fryer.]
“We were pretty surprised,” said Fryer. “We expected perhaps fewer plaques, but to have everything showing up as CAA was unexpected.” When Fryer was a graduate student in David Holtzman’s lab at Washington University, St. Louis, Ron DeMattos had made a similar cross with PDAPP mice and saw no strong redistribution of Aβ to the vasculature (see Jul 2002 conference news). What explains the difference between the models? Fryer told Alzforum that in general, the PDAPP mice have very little CAA, but CAA begins to emerge when the PDAPP/Clu-negative mice age. Fryer has also crossed Clu knockouts with a mouse model of CAA, and vessel pathology runs rampant then as well. “We think this clusterin CAA effect is real,” Fryer said.
Holtzman thought the findings fit with the role of apolipoproteins in the brain. “It is consistent with the fact that clusterin, like ApoE, influences Aβ aggregation and seeding,” he wrote to Alzforum. “Where ApoE promotes CAA, clusterin somehow inhibits its formation.”
What explains Aβ’s dramatic shift to the blood vessels? Fryer suggested two possibilities. The lack of Clu might render the blood vessels stickier, allowing them to trap Aβ. However, since knocking out Clu in a wild-type background caused no obvious change in blood vessels or behavioral phenotype, Fryer favors the second possibility: that without clusterin, ApoE, the other major lipoprotein in the brain, funnels Aβ through the interstitial fluid to the blood vessels, where it gets trapped (see model below).
Wojtas further reported that the Clu knockouts had fewer dystrophic neurites in the brain than APP/PS1 controls. Fryer expected this, since neurites generally become dystrophic around parenchymal plaques. Less expected, however, was that astrogliosis and microgliosis in the knockouts paled in comparison with those in the APP/PS1 brain, and the knockouts spewed less cytokines, suggesting little or no inflammatory response. “We only saw robust gliosis when a few plaques formed, otherwise there seemed to be no inflammation in the knockouts despite a tremendous amount of CAA,” said Fryer.
How do these findings shed light on the association between genetic Clu variants and AD? The single nucleotide polymorphism found in GWAS protects against AD and also seems to increase expression of clusterin. That might suggest that it favors sequestration of Aβ in plaques rather than in the vasculature, but it might also promote degradation of Aβ by microglia (see model above). “We need to look at the APP/PS1 Clu heterozygotes, which more realistically model the human situation where clusterin levels are altered but not absent,” said Fryer. Holtzman noted that human clusterin may act differently to the mouse homolog as well. “That will be important to assess,” he said. Fryer plans to generate humanized CLU mice and develop in vitro assays to see if clusterin alters fibril formation of both Aβ40 and Aβ42, including peptides containing the Dutch and Iowa mutations, which lead to CAA.
Researchers believe that another GWAS hit, TREM2, might also boost microglial clearance of Aβ. However, researchers from Bruce Lamb’s lab at the Cleveland Clinic, Ohio, reported at last year’s SfN meeting that knocking out the TREM2 gene appeared to have little effect on amyloid plaques in the cortices of APP/PS1 mice and even lowered plaque burden in their hippocampi (see Dec 2014 conference news). TREM2 took the field by storm when GWAS identified variants in the gene that almost tripled the risk for AD, making this the strongest genetic risk factor for the disease after ApoE4 (see Nov 2012 news). If this microglial receptor is not required for Aβ clearance, then what about the other major hallmark of AD, neurofibrillary tangles? In Chicago, Shane Bemiller from Lamb’s lab reported that TREM2 helps clear phosphorylated and aggregated forms of tau from mouse brain.
Tau Trouble.
Tau immunoreactivity ramps up in the cortices of htau mice that lack TREM2 (right). [Image courtesy of Shane Bemiller.]
Bemiller crossed TREM2 knockouts with htau mice, which overexpress human tau in place of the endogenous mouse version. These mice express all six isoforms of the human protein and accumulate phosphorylated tau and neurofibrillary tangles as they age. Bemiller reported a huge increase in AT8 and AT180 binding in the cortices of TREM2-negative htau mice compared with htau controls. AT8 and AT180 antibodies recognize various phosphorylated forms of tau and paired helical filaments, precursors to neurofibrillary tangles. AT8 reactivity was mostly in the superficial layers of the cortex, whereas AT180 binding encroached into deeper layers, said Bemiller. In the hippocampus, only AT8 binding surpassed that in control htau mice. However, Gallyas staining revealed that these mice accumulated more NFTs in both the cortex and hippocampus than did controls.
Bemiller concluded that knocking out TREM2 compromises microglial clearance of tau. Interestingly, he found an uptick in the microglial activation marker Iba1 in the vicinity of AT8 immunoreactivity, suggesting that the glia do respond to the tau aggregates but fail to clear them effectively. Other scientists asked if the Iba1 may be on some other cells instead of microglia. Bemiller conceded the possibility, but pointed to the difficulty of identifying peripheral monocytes that enter the brain. Lamb told Alzforum that data from sorted cells and bone marrow chimeras suggests that microglia mediated the effects on tau pathology.
Other evidence suggests that the tau problems in these TREM2 knockouts run deeper than clearance. Phosphorylation of kinases such as JNK, ERK1/2, and GSK3BETA crept up in these mice compared to htau controls, suggesting that hyperphosphorylation of tau might be out of control. Phospho-JNK, normally found in the nucleus, appeared mostly in the perinuclear space in neurons. Bemiller suggested that lack of TREM2 compromises cross-talk between glia and neurons, disrupting cell signaling pathways and causing dysregulation of various tau kinases.
Steve Estus’ lab focused on a different GWAS hit that might also be involved in Aβ clearance. The ATP-binding cassette transporter ABCA7 is found on microglia and may regulate phagocytosis. GWAS uncovered a single nucleotide polymorphism, rs3764650T, which protects against AD (see Apr 2011 news). Estus found that this single nucleotide polymorphism (SNP) increases expression of the protein (Vasquez et al., 2013).
Last March, researchers at deCODE Genetics in Iceland reported a SNP (rs200538373G>C) in intron 41 of the gene that increases risk for AD. That second SNP seems to muck up splicing, causing an extension to exon 41 that introduces a premature stop codon. Scientists at deCODE predicted that rearrangement leads to loss of function (see Mar 2015 news). Estus has looked for the exon extension in tissue samples taken from AD patients and controls. In Chicago, Jared Vasquez from his lab reported that he was indeed able to amplify both the normal and the extended exon forms of ABCA7 from RNA taken from a donor who carried a C allele at that SNP. Surprisingly, Vasquez also found the extended exon in two of six samples taken from people homozygous for the more common G allele. Estus said that some other SNP in the gene may modulate splicing of that exon, though he told Alzforum they have not been able to find a variant that fits the bill. Alternatively, some dietary or lifestyle factors might modulate ABCA7 splicing, said Estus. Vasquez is examining medical records to see if these donors share any history in common that might explain the ABCA7 expression pattern.
Several SNPs near the complement receptor 1 (CR1) gene also associate with late-onset AD (see Jul 2009 conference news). The receptor plays a role in the innate immune system, where it limits complement-mediated damage to host cells. Because it also promotes phagocytosis, researchers believe it may contribute to Aβ clearance. However, interpreting GWAS data has been mired in the biological complexity surrounding this receptor. Red blood cells and peripheral phagocytes make several isoforms, and soluble forms circulate in the plasma. Scientist know less about CR1 in the central nervous system.
Andrea Tenner of the University of California, Irvine, has localized forms of CR1 by immunohistochemistry. In Chicago, she reported that two monoclonal antibodies, 8C9.1 and J3B11, bind CR1 in human brain, and immunoreactivity seems specific to astrocytes. Other labs have detected CR1 on neurons, but Tenner claimed this may be non-specific. In her hands, pre-absorption of antibody solutions with CR1 abolished binding to astrocytes, but not neurons. Neurons may carry non-CR1 epitopes to which the antibodies bind, she suggested. She said it is important for the field that antibody reactivity be carefully characterized.
Using these antibodies, Tenner found that while the extent of CR1 antibody binding varied among different brain samples, no correlation emerged with AD diagnosis or with genotype of the GWAS SNPs, rs6656401 and rs4844609. This suggests that the AD risk alleles do not alter expression of CR1 in the brain. Tenner reported that the risk alleles do slightly increase the affinity of CR1 for their known ligands, the complement proteins C1q and C3b. However, she does not believe this explains the disease association either, because tighter binding would predict better clearance of complement-bound Aβ, which would likely be protective.
One possible explanation, Tenner said, could be poor clearance through the plasma due to lower expression of CR1 on red blood cells. GWAS risk alleles correlate tightly with reduced expression of a long form of CR1 on erythrocytes (see Mahmoudi et al., 2015). In support of this, Tenner noted that less Aβ binds red blood cells in AD patients than in age-matched controls, and that people tend to lose CR1 from erythrocytes as they age (Rogers et al., 2006; Moldenhauer et al., 1988). These observations point to an important role for erythrocyte CR1 in Aβ clearance, but Tenner also noted controversy around the peripheral sink hypothesis. Others at the meeting were intrigued by the data, particularly the finding that only a subset of astrocytes seemed to produce CR1. Some wondered if astrocytes might phagocytose Aβ and how that might be regulated, and if astrocyte expression of CR1 changes with age.
Overall, from the study of GWAS hits thus far, a sense emerged that some of the genes are involved in either clearance of toxic peptides, or their production (see Part 6 of this series).—Tom Fagan
Alzheimer’s GWAS Hits Point to Endosomes, Synapses
In taking a closer look at the Alzheimer’s risk genes enlisted by genome-wide association studies, researchers at this year’s Society for Neuroscience annual meeting, held October 17-21 in Chicago, showed off what confocal and super-resolution microscopy have revealed thus far. They spotted Bin1 and CD2AP in endosomes, and Bin1 in postsynaptic compartments as well. The sightings suggest that both Bin1 and CD2AP regulate processing of the amyloid precursor protein, and that Bin1 also regulates insertion of glutamate receptors into dendritic spines. The work helps scientists better understand how GWAS variants in these genes modify risk for AD (see also Part 5 of this series).
Genetic variants near the Bin1 and CD2AP genes increase risk for AD by about 10 to 15 percent, but researchers know little about the basic biology of these two proteins. Bin1 protein is implicated in endosome recycling. Some prior evidence suggested it might modulate tau, but at SfN, Florent Ubelmann, a postdoc in the lab of Claudia Almeida at CEDOC, NOVA Medical School in Lisbon, Portugal, claimed that Bin1 regulates BACE1 trafficking in axons. In parallel experiments, Ubelmann reported that CD2AP, a modulator of the axon cytoskeleton, helps recycle APP in dendrites. The upshot: Loss of either Bin1 or CD2AP leads to increased production of Aβ.
Ubelmann came to these conclusions after knocking down either protein in mouse wild-type neurons. Aβ ticked up in the axons in the Bin1 knockdowns, while more Aβ dotted dendrites when he silenced CD2AP. To find out why, Ubelmann measured protein trafficking with fluorescent pulse-chase experiments. When he silenced Bin1, BACE1 co-localized more with the early endosome marker Rab5 in axons. The secretase exited much more slowly than normal from endosome tubules, where protein sorting occurs (see image below). APP trafficking was unaffected.
Slow BACE. In wild-type neurons (left), BACE1 (green) exits Rab5-labelled endosomes (purple) within about 30 seconds. Knocking down BIN1 stalls the process (right). [Image courtesy of Claudia Almeida.]
Ubelmann saw something different when he knocked down CD2AP. In these cells BACE1 trafficking appeared normal but, in dendrites, endocytosed APP co-localized more with EEA1, a docking protein on the surface of early endosomes. APP seemed to never quite reach the endosome lumen, which is necessary for its degradation (see image below). Almeida told Alzforum that knocking down either Bin1 or CD2AP increases the chances that APP and BACE1 will cross paths as they traffic through endosomes, leading to enhanced processing of APP.
Out of the Loop. Silencing CD2AP (right) keeps APP (red) out of endosomes (green). [Image Courtesy of Claudia Almeida.]
Researchers at the meeting were impressed by the data but noted that much more needs to be done to understand how the GWAS variants increase risk for AD. The CD2AP data seemed to fit with a recent paper from Fan Liao in David Holtzman’s group at Washington University, St. Louis, showing that N2A cells expressing human APP made slightly more Aβ when CD2AP was silenced. “In this respect, Ubelmann’s data matches what we saw,” Liao told Alzforum. It also fits with an increased plaque burden seen in AD patients carrying the CD2AP risk allele (see Shulman et al., 2013). However, Liao found no change in Aβ pathology when she crossed CD2AP heterozygous nulls with APP/PS1 mice, suggesting that any genetic variation would have to reduce CD2AP levels by more than half to influence pathology. In vivo, microglial CD2AP complicates the picture, since it may influence clearance of Aβ.
Some questioned the relevance of the Bin1 data, since recent work seems to suggest levels of the protein correlate tightly with neurofibrillary tangles in the AD brain and not with amyloid plaques (see Holler et al., 2014). Gopal Thinakaran of the University of Chicago works on BACE trafficking. He told Alzforum that interpreting trafficking experiments can be tricky, as any perturbation to the system can have ripple effects. Thinakaran noted that Ubelmann has not yet shown that Bin1 actually functions in axonal endosomes.
And indeed, in a separate SfN nanosymposium, Britta Schurmann, working with Peter Penzes at Northwestern University, Chicago, painted a slightly different picture of Bin1. Using high-resolution structured illumination microscopy (SIM), Schurmann spotted the protein in the postsynaptic compartment, where it co-localized with the GluA1 AMPA glutamate receptor subunit. This did not surprise Almeida, who told Alzforum that Bin1 likely exists in many cellular compartments.
What does Bin1 do in the postsynapse? Schurmann suggested that it regulates spine morphology. Knocking down the protein in primary cortical rodent neurons, she found fewer GluA1 subunits in dendritic spines and smaller spines. Currents evoked from the neurons were weaker than those from normal neurons. Using SIM to hunt for potential interactions suggested by prior in silico analysis, Schurmann found that Bin1 and the endosome markers Rab11 and Arf6 interact. Silencing Bin1 reduced Arf6 and GluA1 in dendritic spine heads, and Schurmann correlated levels of all three proteins with spine area. She concluded that Bin1 may be a cargo adaptor for GluA1. This could make it important for synaptic plasticity, and its role as an AD risk factor could come to bear via that process, Schurmann suggests.—Tom Fagan
Can Common Genetic Variation in Mice Nail Genes of Aging, Alzheimer’s?
Geneticists have all but exhausted the genome-wide association approach to finding genetic variants that influence AD risk. Massive meta-analyses of more than 50,000 cases and controls have brought the total number of AD risk genes to just more than 20. Can researchers find other AD genes that might be hidden by the cultural and environmental heterogeneity inherent in human populations? Enter the mouse. Mice can be raised in controlled environments. Yet, like people, they have genetic variants that might make them smarter, age better, or, in contrast, render them susceptible to the type of pathology that causes dementia. At this year’s Society for Neuroscience annual meeting, held October 17-21 in Chicago, researchers reported that genome-wide mapping in a panel of inbred mice uncovered variants that accelerated age-related memory decline. The variants lie in the Hp1bp3 gene, which encodes a chromatin-binding protein. Hp1bp3 expression doubles in the hippocampus in AD mouse models and in people who have the disease, said Catherine Kaczorowski, University of Tennessee Health Science Center, Memphis, who presented the data.
The data intrigued scientists at the meeting. “This approach really fascinates me. It may point to pathways and targets [in Alzheimer’s disease] that we have not yet figured out,” said Carol Barnes, University of Arizona, Tucson.
Mouse Diversity.
Panels of isogenic mouse lines may hold key to aging/AD genes. [Image courtesy Catherine Kaczorowski.]
Mouse Panels Capture Genetic Diversity
Kaczorowski found the Hp1bp3 gene by screening the so-called BXD panel of mice. These animals, created by crossing the two common laboratory mouse strains B6 and D2, constitute a genetic reference population that can be studied much the same way as a population of people. The difference is that with the rare exception of identical twins, each person has a unique genotype, while a stable isogenic mouse line represents each genotype within the BXD panel. This allows researchers to study the same genotype multiple times and under varying conditions. There are now more than 150 BXD strains. They have been used to map genetic variants that influence a swath of physiology, including basic metabolism, mitochondrial activity, cardiovascular disease, and longevity (see Koutnikova et al., 2009; Andreux et al., 2012; Hootkooper et al., 2013; Wu et al., 2014).
Kaczorowski moved her laboratory to UTHSC, where Robert Williams had pioneered the use of BXD panels to study complex traits and human disease, so that she could use them to study memory and dementia. Gareth Howell at the Jackson Lab in Bar Harbor, Maine, also uses BXD and other panels of genetically diverse mice to map gene variants that influence pathology in mouse models of AD. Few other labs, if any, are using the approach to study AD. “As far as I know, Catherine and our group are the only ones,” Howell told Alzforum. Other researchers at SfN were unfamiliar with the panel. “The field should really know about these resources,” said Karen Duff, Nathan Kline Institute, New York.
Kaczorowski and colleagues tested 15 BXD strains for long-term memory using a contextual fear paradigm. As graduate student Sarah Neuner outlined in her talk, test scores in middle-aged animals (~14 months old) varied by strain. Neuner mapped the variance to a region on mouse chromosome 4, and Hp1bp3 emerged as the top candidate to explain the variation. The gene is expressed robustly in the hippocampus, and knockouts perform poorly in hippocampal-based working- and long-term memory tests, said Kaczorowski. Neuner found twice as much Hp1Bp3 protein in the hippocampus of 5xFAD mouse models of AD compared to controls; this paralleled a similar increase in Hp1bp3 RNA the hippocampi of 18 women and 15 men with the disease. She used laser capture microdissection of brain samples to determine the transcript levels (see Liang et al., 2008).
How might the gene influence memory? Kaczorowski and colleagues used expression quantitative trait loci analysis to trace the regulation of other genes to the Hp1Bp3 locus. The gene, Wdfy3 (aka Alfy) emerged as a downstream regulatory target of the chromatin-binding protein. Kaczorowski does not know how Wdfy3 influences memory, but it encodes a protein that activates autophagy, a protein-degradation pathway implicated in AD and other proteinopathies (see May 2011 news; Jul 2015 news). In particular, scientists have directly linked Wdfy3 to removal of potentially toxic aggregates of TDP43 and mutant huntingtin (Filimonenko et al., 2007).
Kaczorowski told Alzforum that GWAS identified a locus near the Wdfy3 gene that is nominally associated with AD, but the association did not quite reach genome-wide statistical significance. “An advantage of our approach to identifying disease-modifying alleles is that it reduces the false discovery rate penalty of genome-wide testing, and thereby enhances power to uncover novel risk genes in existing GWAS datasets,” said Kaczorowski.
John Hardy of University College London, questioned whether these mice would be useful to study Alzheimer’s disease. “I think these mice panels will be valuable for dissecting pathways that influence biological processes. I do not think they will be directly useful for helping understand Alzheimer genetics because they do not develop memory loss for the same reason that humans with AD develop memory problems—namely loss of neurons initially in the hippocampus,” he wrote to Alzforum. Howell said that while that might be true, these models can be bred with strains that express AD risk genes and that there are naturally occurring variants within the mouse genome that modify AD genes such as TREM2 and Bin1. Mouse model have been widely used to study AD pathology even in the absence of frank neuronal loss.
Beyond BXD
Kaczorowski plans to combine systems-genetics approaches using the BXD panel with proteomic and other “omic” studies to create network models to better understand and predict memory decline. Kaczorowski and Neuner have already crossed BXD mice with the 5xFAD mouse model of AD in order to identify genetic variants and downstream neuronal mechanisms that mediate individual differences in risk or resilience to AD. They currently have 27 AD x BXD strains and corresponding non-transgenic controls.
Howell plans to use other panels of mice called Collaborative Cross (CC) and Diversity Outbred (DO) to study how genetic variants alter pathology in AD. These mice were developed by a consortium that included JAX scientists. The CC panel are inbred derivatives of an eight-way cross made using different JAX mice, including three strains derived from wild-type mice. To derive DO mice, the researchers outbred the CC mice again. “This gives us hundreds of mice that are genetically unique and provides the levels of genetic diversity present in human GWAS, but without the complicating effects of rare genetic variants and population structure,” said Howell (see JAX site for further information).
Howell first generated a panel of genetically diverse AD models by crossing the APP/PS1 transgenic mice to the founder strains of the CC and DO, and found a range of variable AD phenotypes. He will map genetic changes that associate with that variation using the CC and DO lines. He also plans to use this approach to identify genetic variants that influence late-onset AD genes such as ApoE, TREM2, and other GWAS hits. “The power is that we can do these studies in strains that allow individual genetic variations to be readily identified,” said Howell. The mice can also be used to model the effects of the environment, something that is different for every person included in a GWAS analysis (see Nov 2015 news).
These studies would be difficult to do in a human population. “With the DO panel we can find multiple genetic factors that mimic the complex genetics of human AD,” said Howell. These resources can be used to understand the relevance of GWAS hits to each other, or to identify protective alleles in an unbiased way. “In general, protective alleles are missed by using human GWAS,” said Howell. For example, GWAS missed the protective A673T APP mutation uncovered by whole-genome sequencing (see Jul 2012 news).—Tom Fagan
Preclinical Research Offers New Angles on Immunotherapy
Despite early disappointments, drug developers have bought into immunotherapy for Alzheimer’s disease, banking on a handful of antibodies and active vaccines currently in Phase 2/3 trials and others in Phase 1. As evident from this year’s Society for Neuroscience meeting, held October 17-21 in Chicago, the trend continues. Researchers are still developing immunotherapies for Aβ and for tau, building in new models and strategies to study how comorbidities, such as vascular disease, factor in. Researchers in Italy have even used intrabodies—antibodies produced inside cells—to study the intracellular dynamics of Aβ oligomerization.
Immunotherapy ‘Passifies’ Pyroglutamate Aβ
Most immunotherapies against Aβ have been designed with full-length Aβ1-42 in mind, but some groups have been targeting the truncated Aβ3-x form that undergoes glutamic acid cyclization at the N-terminal. Helen Crehan, who works in Cynthia Lemere’s lab at Brigham and Women’ Hospital, Boston, reported that antibodies to the pyroglutamate (pGlu) form were effective in a treatment paradigm but only if they have good effector function.
Complicating Co-morbidities.
Microhemorrhages (arrows) increase when mice with vascular disease are treated with Aβ antibodies. [Image courtesy of Donna Wilcock.]
Lemere recently published findings that these antibodies can prevent plaques and protect against behavioral deficits when given prophylactically to young APPSwe/PS1ΔE9 mice (Frost et al., 2015). In Chicago, Crehan reported that when given to 12-month-old male mice, which have rampant amyloid plaques, weekly injections of a mouse IgG2a isoform reduced pGluAβ immunoreactivity in the hippocampus at 16 months. Total Aβ and Aβ plaque burden fell as well, indicating that the therapy reduced more than just pGluAβ. “It could be that pGluAβ acts as a seed and we prevented deposition because we removed that seed,” said Lemere. As an alternative, she suggested that microglia activated by the antibody could remove all forms of Aβ. Interestingly, Crehan detected no significant reduction in plaque load when she treated mice with a mouse IgG1 variant of the same antibody. Mouse IgG1 antibodies do not bind Fc receptors on immune cells and therefore have less effector function than their IgG2a homologs. In keeping with the idea that good effector function is necessary for these antibodies to clear Aβ, Crehan reported last March at the International Conference on Alzheimer's and Parkinson’s Diseases in Nice, France, that in ex vivo phagocytosis assays microglia mop up more Aβ when treated with the IgG2a antibody than with any other isoform.
The IgG2a antibody also improved learning and memory. In the water T maze, treated APP/PS1 mice outperformed controls after three months of therapy, though they performed less well than wild-type mice, said Lemere. Nevertheless, Probiodrug, a German company targeting pGluAβ, plans to begin clinical trials of a humanized version of the mouse IgG2a antibody to pGluAβ. Phase 1 trials are slated to begin in 2017.
Active Vaccine for Tau
In a different take on viral delivery, researchers for Kiran Bhaskar’s lab at the University of New Mexico, Albuquerque, described a virus-like particle vaccine targeting pathological phospho-tau. By using chemical cross-linkers, researchers can load empty viral capsids with antigens, and the particle then elicits a strong immune response from B cells. Others have used the strategy for vaccines against Aβ, including Novartis’s CAD-106, which is in Phase 2/3. Bhaskar’s group has used the same Qβ particle to present phospho-tau peptides to the immune system. In her talk, Nikki Maphis said that she can pack about 180 peptides (TPPAPKpTPPSSGQG) from the proline-rich domain of tau onto one virus-like particle.
How well does this work as a vaccine? Maphis tested it in rTg4510 mice, which overexpress human tau with the P301L mutation. They have phosphorylated tau and memory problems at three months and neurofibrillary tangles by about four months of age. Beginning when they were two months old, Maphis gave 10 mice three biweekly injections of p-Tau-VLP or a control particle. After six weeks the mice had generated a high titer of anti phospho-tau antibodies. Maphis detected the antibodies in cortical extracts.
Animals sacrificed at four months old had one-third as much p-tau in their hippocampi as controls and a similar reduction in neurofibrillary tangles in their cortices. The amount of total insoluble tau plummeted in the brain as well. The treated mice mustered fewer CD45+ microglia, indicating less inflammation, and fewer neurons appeared to be apoptotic, suggesting they were protected. The antigen also improved mouse learning and memory. The vaccinated animals did better in a novel-object-recognition test than controls treated with viral particle alone. In the Morris water maze, vaccinated mice behaved much like wild-type, spending about the same amount of time in the target quadrant in a probe test.
Other researchers wondered if the antibodies generated by the vaccine bound tau inside cells, and if so, how much of the antibody actually got to its target. The mechanism is still unclear, but Maphis said that these antibodies could be getting into cells or may be soaking up tau released into the parenchyma, possibly through synapses. Scientists debate whether and how tau spreads transynaptically (see Apr 2015 conference news). Bhaskar told Alzforum that he is looking for partners to help develop this vaccine for the clinic.
Intrabodies for the Endoplasmic Reticulum
In another twist on Aβ immunotherapy, Giovanni Meli from the European Brain Research Institute, Rome, is using intrabodies to prevent cellular damage by oligomeric forms of Aβ. Intrabodies are short, usually single-chain, antibodies that are smuggled into cells under the veil of viral or other vectors (see Jan 2002 news). Meli and colleagues led by Antonino Cattaneo at EBRI have gone a step further, using protein engineering to target the intrabodies to specific subcellular compartments (see Meli et al., 2014). The researchers added a KDEL amino acid motif to the scFvA13 intrabody, which recognizes only oligomeric forms of Aβ in a sequence- and conformation-selective manner. This motif targets proteins to the endoplasmic reticulum. Indeed, Meli found that 7PA2 cells, which express human APP with the V717F mutation, retained scFvA13-KDEL in their ER. 7PA2 cells produce large quantities of Aβ42 and pump various forms of the peptide, including oligomers, out of the cell (see Clearly et al., 2005). Cells expressing the ER-targeted antibody produced 50 to 70 percent less oligomers than did 7PA2 controls, and secreted more monomers into the medium. The research suggested that oligomers can form in the ER and that intrabodies curtail them.
In Chicago, Meli reported that this ER intrabody protects cells from Aβ toxicity. It restored the mitochondrial membrane potential in 7PA2 cells, which supports the idea that AD pathology compromises the integrity of these organelles. ScFv13 also normalized proliferation of neural stem cells from Tg2576 mice when delivered into the cells via lentiviruses. Meli said this intrabody may be useful not only to study the intracellular dynamics of Aβ oligomerization, but also as a potential therapy. He has started in vivo experiments to target the intrabody to the hippocampus of AD mouse models.
Other researchers at the meeting were intrigued by the data but wondered if the intrabody might retain Aβ in the ER, in effect forcing it to oligomerize there. Meli thought this unlikely because he detects no Aβ increase in cells treated with the intrabody.
Passive Immunotherapy and Vascular Dementia
With many vaccination strategies for Aβ and tau in development, do some work better than others for certain patients? Erica Weekman, from Donna Wilcock’s lab at the University of Kentucky, Lexington, addressed this in a mouse study of mixed vascular and parenchymal pathology. A large overlap exists between AD and vascular dementia, such that about 40 percent of AD patients have both morbidities. “Targeting an AD pathological process may be insufficient for clinical outcomes if there are also vascular contributions to the cognitive impairment,” said Wilcock.
To test this in a model, Wilcock’s group fed nine-month-old APP/PS1 mice a diet deficient in folate and vitamins B6 and B12. This drives up homocysteine, causing hyperhomocysteinemia (HHcy), a risk factor for vascular damage. This diet causes vascular cognitive impairment in wild-type mice (see Sudduth et al., 2013). After six months on this diet, the APP/PS1 animals had more microhemorrhages (see image above) and a striking redistribution of Aβ from the parenchyma to the vasculature, though the total Aβ remained the same, said Weekman. The HHcy mice made twice as many errors in a radial arm maze as did the APP/PS1 controls or HHcy alone (Sudduth et al., 2014).
Would this comorbidity affect immunotherapy? Weekman fed nine-month-old APP/PS1 mice the HHcy diet, and three months in, she treated them with weekly intraperitoneal injections of a control IgG or the 3D6 monoclonal antibody—the mouse forerunner of bapineuzumab. The monoclonal elicited a much greater reduction in Aβ in the cortices of mice on the control diet than in mice with vascular pathology. Curiously, 3D6 suppressed microglial activation in HHcy animals, whereas it activated microglia in mice on normal chow. The Aβ antibody held inflammation at bay in the HHcy mice as well, as judged by transcripts for inflammatory markers. While all this data suggested that the antibody might work in animals with mixed pathology, Wilcock believes that the immunotherapy, on top of the amyloid pathology and vascular pathology, may have tipped the innate immune system into senescence. “That’s speculative, but we are looking into it,” she said.
Further analysis indicated all was not well. APP/PS1 mice with hyperhomocysteinemia made more errors in a radial arm maze if they were treated with the antibody, and mouse MRI indicated that the antibody induced even more microhemorrhaging than in untreated controls.
Weekman concluded that anti-Aβ treatment might be unsuitable for people with vascular pathology as a comorbidity to their AD. In fact, ARIA, or Aβ-related imaging abnormalities, a sign of vasogenic edema, contributed to the demise of the bapineuzumab program. To keep edema at bay, dosing in many patients had to be reduced to levels where the antibody was ineffective (see Aug 2012 news). “We still believe that antibody binding to amyloid in the vasculature and the ensuing inflammatory response is the cause of ARIA, but this is still a hypothesis,” said Wilcock. “My guess is plasma HHcy may be a risk factor for ARIA, but that’s pure speculation at this point.”
Wilcock said that it would be valuable to test other antibodies in this mixed-pathology model. She is planning to examine correlations among HHcy, inflammation, AD pathology, and small-vessel disease to get a better handle on how these interact in people.—Tom Fagan
Traumatic Brain Injury—Focus on Heterogeneity, Secondary Damage
Brain injuries often precipitate neurodegeneration, but researchers still know little about the exact mechanisms involved, much less how to treat it. Complicating matters, traumatic brain injury (TBI) can take different forms depending on the nature of the injury, the brain region affected, and how many hits to the head were involved. In recent years, researchers have developed animal models to study these varied types of brain injury. At the Society for Neuroscience annual meeting in Chicago October 17-21, speakers highlighted the distinct pathologies involved in particular forms of TBI, including tau accumulation and axonal injury. Several talks emphasized that the clinical effects of brain injuries vary depending on characteristics such as sex, age, genotype, history, and perhaps even diet. With such a heterogeneous condition, finding treatments is especially challenging. Nonetheless, several researchers described pharmaceutical interventions that preserved neurons and cognition in rodent models when administered soon after injury, hinting that secondary damage can be prevented. Overall, the picture that emerged was of a field still in its infancy, where researchers are just beginning to pin down basic mechanisms by studying a range of animal models.
Myelin Protection. In rats that suffer a brain injury, myelin in the corpus callosum deteriorates (middle) compared to that in controls (left), but treatment with a combination of minocycline and N-acetylcysteine preserves it (right). [Courtesy of Haber et al., 2015, submitted.]
Epidemiological studies indicate that head injury confers a roughly 60 percent increased risk of dementia (see Aug 2012 news; Jun 2014 news). But what type of dementia? Some evidence suggests that a single severe blow to the head pumps up amyloid production, and may predispose people to Alzheimer’s (see Nov 2013 news; Jan 2014 news). On the other hand, repetitive but milder injuries, such as those suffered in contact sports or from proximity to explosions in the military, trigger a progressive tauopathy known as chronic traumatic encephalopathy (CTE) (see May 2012 news; Nov 2012 news series; Mar 2014 news). In particular, these repetitive injuries seem to shred fragile axonal connections (see Jan 2013 news).
Many Mechanisms Underlie TBI
Because of this high-profile CTE research, head injuries became synonymous with tauopathies in many researchers’ minds, noted Fiona Crawford of the Roskamp Institute in Sarasota, Florida. However, she and other speakers stressed that TBI is a diverse condition, and its study requires many different animal models of injury. Researchers have now developed protocols that mimic single severe blows to the head, repetitive mild impacts, and explosive blasts, among others. These models vary widely in how much tau they accumulate, as well as in other features, suggesting they may capture the heterogeneity of the syndrome. However, researchers at SfN noted many points of convergence between different forms of injury as well.
Many researchers use a controlled-cortical-impact model, in which a piston strikes either the skull or exposed brain of a rodent. Crawford’s group found that mice that received five mild, closed-head controlled cortical impacts developed neuroinflammation and axon damage along with progressive motor and cognitive defects, but no changes in tau (see Mouzon et al., 2012; Mouzon et al., 2014). Apparently, injured brains do not need to develop tau pathology to experience long-lasting, progressive damage. “It’s not all about tau,” she concluded.
Nonetheless, to study the role of tau in CTE, Crawford developed a protocol to induce chronic TBI. She exposed mice that expressed wild-type human tau to two head injuries per week for two to three months, mimicking an athlete exposed to frequent concussions. In this model, both total tau and phosphorylated tau rose, persisting for at least three months after injury, she reported. In addition, neuroinflammation spiked, cerebral blood flow dropped, and the animals had trouble learning. These mice may model CTE-like degeneration, she suggested.
TBI Beyond Tau
Overall, few of the talks at SfN focused on tau, instead highlighting other aspects of brain damage. Michal Vascak in John Povlishock’s group at Virginia Commonwealth University, Richmond, detailed what happens to axons after brain injury. Vascak used a model of mild TBI in which a device rapidly injects a small volume of saline into a mouse brain, subjecting cells to a fluid concussion wave. This does not cause brain lesions or hemorrhages, but a diffuse, widespread axonal injury ensues as fragile neuronal connections twist and shear around the injury site. Moreover, even those axons that do not break may be affected, Vascak said. He reported that two days after injury, intact axons did not fire properly. He wondered if this might be due to changes in the axon initial segments, where action potentials are generated.
To get a closer look at those segments, Vascak used confocal microscopy to image individual uninjured axons in postmortem mouse brain two days after injury. Using specific markers to identify the ends of initial segments, he found that the distal end had shrunk by about 2 μm. Since this end triggers action potentials, the change would alter neuronal firing properties, and that in turn might affect overall network excitability, Vascak suggested. The data demonstrate that TBI can affect the properties of even intact axons.
How does this happen? Perhaps disconnection of upstream neurons caused by the TBI shifts axon initial segments to a malleable, dynamic state, Vascak hypothesized. This might allow a localized injury to have widespread effects on brain function, as broken axons lead to changes in downstream neurons, which then affect neuronal activity even further downstream. The data jibe with a similar finding of shortened axon initial segments in a blast model of traumatic brain injury (see Baalman et al., 2013). There is precedent for axon initial segment plasticity. They change in length and position in response to alterations in neural activity (see Grubb et al., 2011).
In addition to axonal damage, brain injuries are marked by the pathogenic accumulation of proteins, such as Aβ and TDP-43 in addition to tau. Chinmoy Sarkar, who works in the lab of Marta Lipinski at the University of Maryland School of Medicine, Baltimore, proposed a single mechanism that could explain all these pathologies. The researchers previously reported that a controlled cortical impact disrupted autophagy in cortical and hippocampal neurons. Lysosomes lost the ability to chew up unwanted proteins, and autophagosomes accumulated (see Sarkar et al., 2014). At SfN, Sarkar reported that a rise in cytosolic phospholipase A2 (cPLA2) a few hours after brain injury disrupts autophagy. The phospholipase breaks down lipids in lysosomal membranes, allowing proteases such as cathepsin D to leak out into the cytosol. This limits proteolysis in lysosomes, creating an autophagy roadblock. Treating mice with an inhibitor of cPLA2 after TBI prevented the accumulation of autophagosomes, he said.
Same Injury, Different Outcomes
Even as speakers detailed the multiple things that can go wrong in injured brains, they also stressed specific effects that depend on the characteristics of the animal. In an SfN press conference, Ramesh Raghupathi of Drexel University College of Medicine, Philadelphia, reported that the brains of males and females respond differently to TBI. When mice were subjected to a forced-swim test one to two months after a mild brain injury, males gave up swimming sooner than females. This is typically interpreted as a sign of depression. In addition, dopaminergic transmission fell in the males, another sign they were depressed since dopamine increases the sense of well-being. Females, on the other hand, reacted more strongly than males to a light touch on the face, suggesting they felt more pain. Both depression and headache are common symptoms in people in the months after brain injury. It is not yet clear what determines these sex differences, Raghupathi said.
Age is another crucial factor, with children often having worse outcomes after brain injury, including more post-traumatic seizures than adults (see Arango et al., 2012). At the press conference, Trent Anderson of the University of Arizona in Phoenix reported that three-week-old mice, but not adults, developed epileptiform brain activity after a single moderate injury to the exposed brain (see Nichols et al., 2015). In the brains of these pups, Anderson saw an 80 percent drop in levels of parvalbumin, which marks a subclass of inhibitory interneuron. The neurons themselves did not die, but they changed in shape and size, suggesting they might be disconnecting from the network, Anderson said. The removal of these inhibitory neurons likely amps up overall network excitability. This may explain the greater susceptibility of children to seizures after brain injury, he speculated.
Genotype can also be important. Carriers of the ApoE4 allele have up to 10-fold higher risk of dementia after brain injury, for example (see Mayeux et al., 1995; Zhou et al., 2008; Zeng et al., 2014). At SfN, Crawford reported data from a small study comparing mice that carried the human ApoE4 or ApoE3 alleles. The E4 carriers produced significantly more phosphorylated tau after injury, she said. Crawford also described preliminary data suggesting that mice eating a high-fat, high-carb “Western diet” fare worse after TBI than those consuming healthy, low-fat chow.
Could life experiences also prime the brain to be vulnerable to TBI? Sydney Candy, working in Quentin Pittman and Michael Esser’s labs at the University of Calgary, Canada, found that to be the case. She injected 10-day-old rats with either saline or the inflammatory agent LPS to mimic an early life infection. Twenty days later, the rats received a mild controlled cortical impact brain injury. Rats that had been primed with LPS performed more poorly on novel-object-recognition tests, failed sooner in the forced-swim test, and spent more time in the center of open areas in their cages than did control-injected littermates. In control rats, cytokine levels surged after TBI, but in the LPS-primed animals, they remained at wild-type levels, suggesting failure of an immune response. The data hint that prior neuroinflammation could make the brain more vulnerable to the effects of injury, Candy said.
Neuroprotective Strategies
With such a diverse array of injuries and effects, what are the options for treating TBI? Researchers at SfN suggested targeting neuroinflammation and oxidative damage, which are common after brain injury. Michael Ayo Sangobowale in Peter Bergold’s lab at SUNY Downstate Medical Center, Brooklyn, reported that a combination of the antibiotic minocycline and the modified amino acid N-acetylcysteine (NAC) might help. Both of these drugs have antioxidant and anti-inflammatory actions, and also modify gene expression; it is not clear which of these actions provides the therapeutic benefit, Bergold wrote to Alzforum. Previously, Bergold and colleagues reported that the two drugs together better preserved memory than either alone when given to rats one hour after a mild injury to the exposed brain. The combination prevented neuroinflammation and preserved the myelin sheath around axons (see image above; Abdel Baki et al., 2010; Haber et al., 2013). At SfN, Sangobowale extended this data, presenting evidence that the drug combo protects myelination even when given 12 hours after the initial injury. He is currently investigating how long the therapeutic window lasts.
In addition, Sangobowale reported that rats dosed 12 hours after injury performed better than untreated controls on an active place-avoidance test, in which animals have to learn a pattern of cues in order to avoid a spot where they will receive an electric shock. Treatment 24 hours after injury did not improve performance on this test, but did help rats do better in a Barnes maze, in which animals have to escape down a hole in a set location. Active place avoidance demands a higher level of cognition than the Barnes maze, Bergold wrote to Alzforum. “Therapeutic windows may differ depending upon what outcome measure is being assessed,” he noted. But overall, the take-home was that early treatment has better outcomes.
Many groups are interested in the potential of hyperbaric oxygen therapy to treat the injured brain. In this approach, patients breathe 100 percent oxygen at high pressure for a short period each day. The extra oxygen is believed to nurture and preserve injured neurons, but small clinical trials have yielded mixed results, with one review concluding that more data are needed to determine what type of injuries will benefit from this approach (see Bennett et al., 2012). At SfN, Chaim Pick of Tel Aviv University, Israel, reported that the therapy prevented cognitive decline in mice. One week after receiving a blow to the side of the head, control mice learned poorly in a Y-maze and failed to recognize novel objects as new. However, mice that received hyperbaric oxygen therapy for one hour per day, starting three hours after injury and repeated for four consecutive days, learned as well as wild-types. Injured controls had lost about 20 percent of their neurons and 40 percent of their myelin, while displaying 25 percent more reactive astrocytes. The brains of treated mice, on the other hand, looked like wild-types. The results provide clues as to how oxygen treatment may help damaged brains, the authors suggested.
Bruce Citron at Bay Pines VA Healthcare System, Florida, highlighted a key role for antioxidants. Previously, he had reported that expression of the transcription factor Nrf2, which turns on antioxidants and chaperones, ramped up after a closed-head injury in mice. Treatment with an activator of Nrf2 improved memory in the injured animals, suggesting that Nrf2 upregulation represents a protective response to injury (see Saykally et al., 2012). At SfN, Citron extended these data to a blast-injury model developed by Pick. In collaboration with Jessica Chang at Bay Pines, Citron exposed mice to a pressure wave from an explosive blast to simulate injuries experienced by military service members. As in the impact model, Nrf2 expression soared. Mice treated with an activator of Nrf2 once daily starting 30 minutes after a blast injury expressed less amyloid precursor protein and neurodegenerative markers than controls, Citron said. “There seem to be common themes as well as differences between different types of injuries,” he wrote to Alzforum. Citron is looking for additional ways to modify gene expression after TBI to protect the brain, he added.
Boost in calcium flux after the initial insult might also contribute to pathology, said Rachelle Dugue of SUNY Downstate Medical Center. Dugue works with Douglas Ling at SUNY and Jeffrey Goodman at The Institute for Basic Research in Developmental Disabilities, Staten Island, New York. Increased calcium activates the protease calpain, which cleaves numerous substrates and precipitates neurodegeneration, Dugue said. She evaluated the ability of a new calpain inhibitor, ALA-1.0, to prevent this. ALA-1.0 consists of the known calpain inhibitor leupeptin linked to the anti-epileptic drug pregabalin, which helps the whole compound to cross the blood-brain barrier. When Dugue gave rats 80 mg/kg ALA1.0 immediately after a controlled cortical impact, then examined them two days later, 40 percent fewer neurons degenerated than in controls. In ongoing work, Dugue will determine the therapeutic window for ALA-1.0, and whether the drug also preserves learning and memory.
While overall the presentations at SfN showcased a fledgling field, researchers were energized by the progress made thus far, by the many opportunities for future study, and by the potential to turn some of the discoveries into therapeutic interventions. In particular, scientists agreed that the ability to model the different types of brain injury has helped propel the field forward.—Madolyn Bowman Rogers







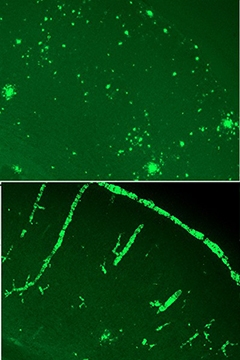
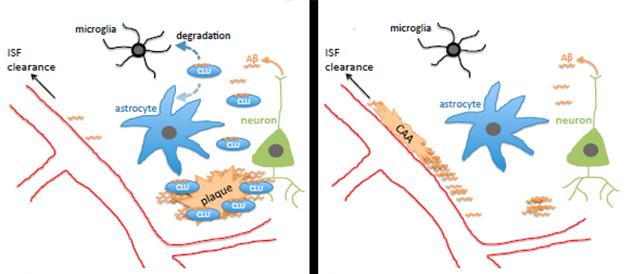
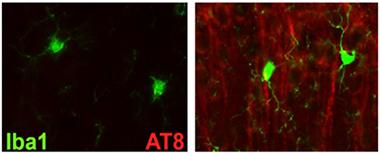
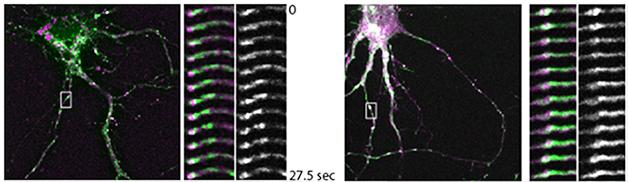




Comments
No Available Comments
Make a Comment
To make a comment you must login or register.