First results from the Genetic FTD Initiative, and the advance of an HDAC inhibitor drug into Phase 2, made big splashes at the ICFTD conference held last month in Vancouver. Five hundred and ninety scientists from 30 countries met to exchange the latest clinical and scientific news on the diseases that make up FTLD. Multi-center cohort studies in Europe and North America reinforced the sense that there is a community spirit that may allow a common approach to how to develop therapies. Frontotemporal lobar degeneration can start in myriad ways—with social disinhibition, overeating, halting speech, or even odd misperceptions of pain or cold—but it always ends in dementia. Attendees said that the FTD field is poised for rapid progress.
Vancouver Frontotemporal Dementia Conference Shows Awakening Field
It can start with insensitive comments at the dinner table. It can start with theft by a previously law-abiding citizen, or sexual misconduct by a hitherto faithful spouse. It can start with halting speech or a blank stare at a simple sentence. No matter the beginning, frontotemporal lobar degeneration (FTLD) usually ends in the mute, bed-bound misery of advanced dementia, and death.
This mysterious set of neurodegenerative diseases was the topic when 590 researchers from 30 countries met in the rainy city of Vancouver, Canada, on October 23 and descended into a hotel basement ballroom for 2½ days of exchange about the latest research findings. When they re-emerged, into a still-rainy city, they expressed awe at how their branch of study has hurtled forward in the eight years since the rapid-fire discoveries of progranulin, TDP-43, C9ORF72 hexanucleotide repeats, and tau PET tracers essentially broke it open. “The field has built on these discoveries so rapidly and with such volume, it’s remarkable,” said Ian Mackenzie of the University of British Columbia.
Co-hosted by Mackenzie and Howard Feldman of UBC, the 9th International Conference on Frontotemporal Dementias (ICFTD) featured 56 talks, 319 posters, and 13 satellite meetings—a far cry from its humble beginnings in 1986 in Lund, Sweden. FTD research languished for years in the shadow of its larger cousin, Alzheimer’s research, but now the pieces have moved into place for it to draw broad-based interest among scientists, funders, and even some drug-development companies. In fact, many players from the Alzheimer’s field are pushing in, sensing action and the chance to find something big. Clinical, neuropathological, and molecular genetics studies are each advancing. A detailed classification system is gradually being worked out; it is heterogeneous and therefore complex, but serves as a basis from which to work, scientists agree. Cellular pathways from lysosomal storage to RNA regulation are coming into view. Therapeutic trials are starting to target underlying mechanisms.
This diffusion tensor image shows fiber tracts of the speech-production network that degenerate in primary progressive aphasia, a language form of FTLD. The orange tracks, for example, connect the supplementary motor area to the Caudate nucleus. [Image courtesy of Maria Luisa Mandelli and Marilu Gorno-Tempini, UCSF. For more, see Mandelli et al., 2014.]
Perhaps the most important innovation at this moment lies in newly forged international collaborative relationships to comprehensively examine patients and their families in longitudinal cohort studies. Inspired by the success the Alzheimer’s field has had with natural history cohorts such as the Alzheimer's Disease Neuroimaging Initiative (ADNI), the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL), and the Dominantly Inherited Alzheimer’s Network (DIAN), every FTD center participating in such a joint venture agrees to use the same methods of deep phenotyping, genotyping, and imaging, and to pool their findings into shared databases. At ICFTD, two such initiatives presented baseline data, one on 336 presymptomatic mutation carriers in Europe and Canada (see Part 2 of this series) and one on 700 symptomatic patients in Germany (see Part 3). Two large North American initiatives announced that they had just received $5.1 million to get going on their own cohorts (see Part 4). “Working across the progranulin, C9ORF72, and tau forms of these diseases is an accessible problem, but it requires global effort,” said Feldman. “There is tremendous power when we work together.”
Therapeutic studies are beginning, as well. Here, researchers work amid two cross-currents. They talk about avoiding the mistakes that have marked the history of Alzheimer’s trials, where some drugs were rushed into Phase 3 without sufficient evidence that they entered the human brain and adequately engaged their molecular target there to have a chance at making a dent in the disease. At the same time, a sense of urgency to relieve the profound suffering FTD causes pervaded this conference. This awareness was made more acute by a simultaneous meeting of 200 FTD caregivers hosted by the Association for Frontotemporal Degeneration (AFTD). Most of the carers had traveled to ICFTD from outside British Columbia. “In FTLD there is a faithful network of caregivers that spans the globe,” Feldman said. “Caregivers hold the key to what matters; we need to listen to them.”
No one disputes that new therapies that target central FTLD molecules are sorely needed. No drugs approved specifically for FTLD exist. Rather, some patients with behavioral symptoms take antidepressants with modest success; patients with parkinsonian symptoms often are prescribed levodopa, but typically do not respond to it.
As scientists tried to advance the understanding of frontotemporal dementia from basic science to clinical trials, their discussions reflected the realization that they are working together through the early days of translational medicine, rather than entrenched in competing interests. “We heard people from different centers and backgrounds stating similar viewpoints about how to go about those trials. We have not just random individual strategies; there really seems to be a community approach to this,” said Mackenzie. “That gives me optimism that the translational process will be effective.” Throughout the conference, scientists showed unpublished data on work in progress. They asked colleagues from other labs to pressure-test a new assay, or offered to teach them new analysis algorithms. “There is a spirit of generosity right now where people are forthcoming with their assets to work toward a common goal. That touches us all as a community,” Feldman said.
The Clinical Picture
Some 50,000 people in the United States have some form of FTD. The pathological name, frontotemporal lobar degeneration (FTLD), is an umbrella term for a diverse set of diseases that are all marked by atrophy starting somewhere in the frontal or temporal lobe of the brain, often on one side. Beyond this common feature, however, FTLD is heterogeneous at every level—the clinical presentations, the underlying neuropathology, the neural networks that become dysfunctional, and the genes that cause the havoc. Symptoms, proteins, and genes do not map neatly onto separate pathways, but into overlapping groups. Researchers now agree that FTLD represents a spectrum of diseases that stretches toward parkinsonian symptoms one on end and amyotrophic lateral sclerosis (ALS) on the other.
Outwardly, frontotemporal dementia comes in two main flavors, though subtypes and atypical variants abound and not every research center groups them in exactly the same way. In defining the expressions of FTLD, clinicians are redrawing the line between psychiatry and neurodegeneration. The signature disease is behavioral variant FTD, or bvFTD. This starts primarily as a disease of social cognition, emotion, and executive control, said Bruce Miller of the University of California, San Francisco. A formerly warm, interested, disciplined person can become selfish, apathetic, and gluttonous. He may ignore distress in others, or do embarrassing things in public. She may abuse drugs or alcohol, or become compulsively engaged in the arts or business. Misdiagnosis happens in both directions: bvFTD is frequently mistaken for depression or schizophrenia, but people are also referred to UCSF with a diagnosis of bvFTD when in fact they have a psychiatric illness. “Apathy, change in eating habits, and disinhibition are great distinguishing factors between psychiatric disease and bvFTD that are not yet being applied in the diagnostic community,” Miller said.
The other main form of FTLD, primary progressive aphasia (PPA), starts with problems understanding speech (the semantic variant) and generating speech (the nonfluent or agrammatic form). PPA starts with specific deficits, such as confusion about the meaning of words and concepts, or halting, laborious speech. The underlying functions map to frontal cortical and subcortical areas and the left posterior insula. They are connected into language networks by fiber bundles, such as the aslant tract, that are the subject of study with diffusing tensor imaging and tractography (see image above). At ICFTD, Marilu Gorno-Tempini of UCSF discussed emerging evidence to support a suggestion proffered originally by Marsel Mesulam at Northwestern University, Chicago, that certain developmental language disabilities may render the brain’s language network less resilient. Childhood dyslexia, for example, may make the brain more prone to a particular, logopenic subtype of PPA later in life.
In addition, neurologists record a long and growing list of atypical frontotemporal dementias. The motor symptom variant corticobasal degeneration (CBD) renders the upper body rigid and the arms increasingly unusable until the hand becomes permanently clenched in a fist, said Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. Less well known is a language variant that starts with what is called prosopagnosia, a curious inability to recognize the faces of familiar people. Patients with this disease will view a photo of Barack Obama and say “I like him” or “I do not like him,” but will not be able to name the president. Boeve told the story of a man who played bingo with his second wife. The man stepped away, chatted with someone, and when he returned his wife asked, ”Why were you talking to her?” He replied that he just had a talk with someone—he did not realize the person was his first wife. Another rare variant is primary progressive apraxia of speech. Disease in these people starts with reversals of the correct response. “Are you married?” “No! .... Yes.” Clinicians agreed that the heterogeneity of FTLD subtypes urgently demands robust biomarkers to help them diagnose what causes a given patient’s disease.
At ICFTD, John Hodges of Neuroscience Research Australia in Sydney discussed the puzzling phenomenon of FTLD phenocopies. A minority of bvFTD and aphasia patients meet diagnostic criteria when they come in, but then stay stable for many years (Rascovsky et al., 2011; Khan et al., 2012). This runs counter to the classic view of neurodegenerative disease as inexorably worsening. It is good news for the individual patient, because it means not everyone who receives a bvFTD diagnosis will head toward profound suffering and death in only a few years. These patients can be bad news for clinical trials, however. If researchers unwittingly enroll a “phenocopy” soon after diagnosis, they might mistake his lack of progression for a drug benefit.
At ICFTD, Emma Devenney in Hodges’ group noted that six-year observational data tipped them off to certain clues that predict a suspected bvFTD patient is not a phenocopy, but will indeed progress. The clues include a family history, low baseline performance on memory, visuospatial- and language-based tests such as the RAVLT, poor self-care or other activities of daily living, and definite atrophy on MRI. “It makes sense to think that if a person has at baseline a deficit in a domain we know is part of the spectrum, such as memory, then it is more likely to be an organic disease and not just an isolated personality problem that may phenocopy bvFTD,” commented Mackenzie. Some slow progressors who did eventually get worse after an initial stable phase turned out to have C9ORF72 mutations, Hodges said.
The Genes and Proteins
Beneath the panoply of outward expressions of disease lie three core RNA/protein pathologies, but they do not map in one-on-one relationships to a given clinical type. A few subtypes of FTLD do have a consistent underlying pathology; for example, semantic dementia patients typically have TDP-43 pathology in their brain, and progressive supranuclear palsy patients have tau pathology. But the majority of FTLD cases could be driven by any of a number of different underlying neuropathologies; “guestimating” the correct one has become part research, part art form. This is true of bvFTD, as Bill Seeley at University of San Francisco and collaborators documented on a poster at ICFTD. At least six different protein pathologies have been described for this clinical syndrome. Likewise, tau, TDP-43 and even Alzheimer’s pathology underlie the primary progressive aphasia syndromes in about equal measure, said Mesulam.
FTLD is highly genetic. Up to 50 percent of all cases have a family history, and 10 to 30 percent are autosomal-dominant. Mutations in the genes for tau, progranulin, and C9ORF72 account for a majority of familial cases, but more genes continue to be implicated in FTLD overall. Neurogeneticists disagree about whether additional highly penetrant genes remain to be found. At ICFTD, geneticists proposed several new genes that may alter FTD risk or change the course of the disease in people who already harbor one of the dominant mutations. Christine van Broeckhoven of the University of Antwerp, Belgium, proposed VPS13C as a new FTD gene. Rosa Rademakers of the Mayo Clinic in Jacksonville, Florida, found that expression of hox genes was boosted in C9orf72 carriers. Hox (aka homeobox) genes were identified as master regulators of patterning during development, but they also play a role in neuronal repair. Studying a family with FTLD in whom she had excluded all the known dominant mutations, Rademakers presented to the audience a list of potential genetic variants she thought might be responsible for this family’s disease, and asked the scientists to dig through this list of genes in their own unexplained familial samples in hopes of homing in on a new genetic culprit (see Part 8 of this series).
Mapping from gene to neuropathology is more straightforward than mapping from pathology to clinical syndrome. Mutations in the gene MAPT give rise to tauopathy; mutations in progranulin, C9ORF72, and VCP give rise to TDP-43 pathology. Alas, tau and TDP-43 pathologies each can lead to a range of different and overlapping clinical pictures, even in a single family. Mutations in the gene FUS cause a different neuropathology and lead to bvFTD or ALS (for review, see Riedl et al., 2014). In Vancouver, a key focus was on presentations devoted to parsing the molecular mechanisms of how the pathogenic hexanuceotide repeats in C9ORF72—in the form of RNA foci, peptide inclusions, or an unknown soluble entity—relate to aggregates of TDP-43, and which part of this axis of evil ends up killing neurons (see Part 5 of this series).
Even as this picture was being filled in, researchers at ICFTD saw a problem in the wide span of symptoms that can develop from each gene or protein a given therapy might target. This range will make it difficult for symptom-based outcome measures to capture whether the drug works. Early stage trials can start out by addressing just the question of whether the drug changed levels of its immediate target, e.g. progranulin, as their efficacy outcome (see Part 6 of this series). But where to go from there, asked Feldman? How to tell whether elevating progranulin levels benefits a treatment group?
On this question, the conference showcased budding research on physiological markers. They could, if they hold up in further research, be developed into “intermediate” outcome markers that lie downstream of target engagement, but remain short of classical clinical symptoms of FTLD. For example, it appears that people with FTLD have abnormal perceptions of pain, temperature, or even music. Researchers working with Jason Warren at University College London, U.K., are characterizing brain-network signatures that could explain why a bvFTD patient can wear a fur coat on a hot day, yet remain unaware of her overheating body and the beads of sweat standing on her forehead. The conference also featured news on social cognition. Researchers are fingering some of the same brain networks that have been implicated in the personality changes seen as disease progresses and formerly warm personalities become emotionally blunted (see Part 7 of this series). Readers used to the entorhinal cortex, perforant path, and hippocampal pyramidal neurons: Get ready to hear about the insula, thalamus, uncinate fasciculus, and von Economo cells.—Gabrielle Strobel
First Data from GENFI1: Brain’s Insula Region Shrinks A Decade Before FTD
At the 9th International Conference on Frontotemporal Dementia (ICFTD), held October 23 to 25 in Vancouver, British Columbia, Canada, the biggest news splash happened when the first international cohort study of presymptomatic FTD mutation carriers presented its initial results, on brain volume and neuropsychological tests. In a nutshell: It appears as if each causative FTLD gene generates its own particular sequence of events as it ravages the brain, but overall atrophy eats away the insula first. Folded beneath the fronto-lateral surface of the brain, this area of the cerebral cortex is a connectivity hub of the salient network, involved in the regulation of consciousness and emotion, and the body’s homeostasis. The news came as validation but not surprise, as the insula has lit up previously in small studies of presymptomatic FTD gene carriers, as well as in studies of early symptomatic FTD. (Thirty presentations at ICFTD mentioned the insula.) In the paper-and-pencil department, deficits in behavior and cognition began to show five to 10 years after brain atrophy, a few years prior to the estimated age of onset.
GENFI will be entering Phase 2 in spring 2015.
Jonathan Rohrer of University College, London, presented results of a first data cut in the Genetic FTD Initiative (GENFI), a 13-center study of carriers of pathogenic mutations in tau, progranulin, and C9ORF72. The work generated respect throughout the conference. “This study is super-important,” said Markus Otto of the University of Ulm, who coordinates a large cohort of symptomatic FTD in Germany (see Part 3 of this series). “GENFI did spectacular work,” said Adam Boxer of the University of California, San Francisco, whose center just nabbed NIH funding to do much the same thing in the United States and other Canadian sites (see Part 4). “Impressive data,” ICFTD co-host Howard Feldman said in his concluding remarks.
Set up in 2011, GENFI is a consortium of centers in Canada, France, Germany, Italy, Portugal, Spain, Sweden, the Netherlands, and the United Kingdom. Martin Rossor and Rohrer at UCL jointly coordinate the network. GENFI is similar to DIAN, the Dominantly Inherited Alzheimer's Network (Nov 2008 news story), in that it aims to chart the evolution of FTD in the decade or more before a person shows symptoms. The idea is to use the autosomal-dominant forms, in which researchers know that a mutation carrier will develop the disease, to learn whether there is a measurable, stereotypical sequence of brain changes that precede the symptomatic phase. If there is an ordered pattern of biomarker change, then these stages can be characterized by comparing mutation carriers to their non-carrying siblings in longitudinal natural history studies. In so doing, GENFI can—as DIAN did—establish a so-called “readiness” cohort of people whose presymptomatic biomarker changes are known and who want to enter therapeutic intervention trials before large swaths of their brain are damaged beyond repair. “Our goal is to create a global initiative for therapeutic trials. Our ultimate goal is to provide a disease-modifying treatment for genetic FTD,” Rohrer said.
For the first two years, the investigators’ goal was to see if they could pull off GENFI. Would they be able to recruit enough participants to build a sizable cohort? After all, FTD is a rare, heterogeneous disease that inflicts both daily dysfunction and fear of inheritance on affected families. Would they agree on standardized assessments? Could they harmonize sample collection and data handling in a uniform way across centers, countries, and languages? Researchers like to do things their own way, but in consortia they have to give up local control on many issues in service of a greater good. They have to adhere exactly to centralized methods for cerebrospinal fluid processing, and perhaps forgo their favorite imaging techniques for ones that hold up more robustly in multicenter settings. They may have to map their data to new standards, fork over results to a common database, and, on manuscripts, drop their name into a large collective of authors. It worked for GENFI. To date, GENFI has enrolled 336 people and 99 have returned for their first follow-up visit. Setting up a standardized protocol was key, Rohrer said.
GENFI welcomes asymptomatic carriers of mutations in tau, progranulin, and the C9ORF72 gene, as well as their mildly symptomatic and mutation-free siblings. Most people prefer not to know their mutation status, Rohrer said. GENFI protects their privacy by having “genetic guardians” enter the data into a database so that clinicians who interact with the participants do not see it and cannot accidentally deduce or disclose whether a given person has the mutation.
Like DIAN, GENFI asks a lot of its participants. They sit for a long battery of tests measuring their social cognition, executive function, memory and other domains of mental function. They submit to lumbar punctures for CSF sampling, blood draws for plasma biomarker research, and lie in a scanner for volumetric, functional, and structural connectivity MRI. Their least favorite assessment? “The neuropsych tests. They do not mind the scanner or the spinal tap,” Rohrer said. The researchers try to help by explaining that the tests are hard, designed to trip up healthy people. Still, these test sessions can be daunting to a person who has grown up watching FTD in a parent and now confronts his or her own risk head-on during each visit.
In September 2013, GENFI researchers froze the data to generate a first batch for analysis. By that time, 220 people had finished their first visit. Of those, 118 are from progranulin, 44 from tau, and 58 from C9ORF72 families. GENFI started just before the C9ORF72 repeat expansion was published in September 2011, hence this first cut reflects a preponderance of people with progranulin mutations. More C9ORF72 families have since joined, Rohrer said. Forty people are symptomatic; 45 are presymptomatic carriers of progranulin, 15 of tau, and 18 of C9ORF72 mutations; 102 are non-carriers. “We would expect in autosomal-dominant disease a 50-50 split between carriers and non-carriers, but we have over-representation of the latter,” Rohrer said. He does not know why, but Boxer, who studies presymptomatic FTD at the University of California, San Francisco, said the same is true at his site.
Comparing carriers and non-carriers, the scientists looked for links between a given measure and the expected time of symptom onset in each carrier. In GENFI this is calculated as the difference between the participant’s age and the average age of onset in their family.
When did the first cognitive signals appear? Carriers and non-carriers started to diverge around five years before estimated onset, though the specific tests were not the same from gene to gene. For tau and C9ORF72, the Cambridge Behavioral Inventory CBI-R showed the first differences; for progranulin, it was executive function and working memory.
Before even these subtle signs appear, the brain appears to have been shrinking for a decade. GENFI measures the volume of frontal, temporal, parietal, occipital, insular, and cingulate cortex, as well as the subcortical areas of the hippocampus, amygdala, thalamus, and striatum. The first difference between carriers and non-carriers was in the insula, at 15 to 10 years prior to estimated onset. In roughly five-year steps, the temporal lobe began to atrophy, then the frontal lobe, then the parietal lobe. The cingulate cortex changed around onset of symptoms and the occipital lobe soon after, Rohrer reported.
Unlike in Alzheimer’s, where three autosomal-dominant genes converge on APP processing, the three main FTD genes are not known to act on a common pathway, and no single pathogenic hypothesis has been formulated. GENFI analyzes all results separately by gene, and is finding a unique order of events for each. At ICFTD, Rohrer showed that atrophy in tau carriers appears to start in the hippocampus 20 years before onset, spreading to the amygdala and temporal lobe. Atrophy in progranulin carriers is first seen in the insula at minus 15 years, then in the temporal and later the parietal lobe; the first subcortical target is the striatum. In C9ORF72 carriers, early atrophy is most widespread, cropping up 20 years before estimated onset in the insular and parietal cortex, hippocampus, and thalamus. In this latter region, the loss of volume is most pronounced at that early stage, Rohrer said. These numbers and sequences will be refined as data from the second cut, taken in October 2014 on 336 participants, are analyzed.
The first cut of follow-up data, on 99 participants, came in for analysis just before ICFTD. A quick peek suggests differences in how fast the brain as a whole shrank in the year between non-carriers (0.1 percent), presymptomatic carriers (0.6), and symptomatic carriers (1.4), Rohrer said. Most of that analysis remains to be done.
Several scientists noted that the estimated of date of onset is just that, particularly for the C9ORF72 repeat expansion whose age at onset ranges widely. At ICFTD, Christine van Broeckhoven, University of Antwerp, Belgium, reported “anticipation” in some families with C9ORF72 hexanucleotide repeat mutations, confirming previous reports (e.g., Chiò et al., 2012). In this strange phenomenon known from the trinucleotide-repeat disease Huntington’s, subsequent generations become symptomatic at younger ages than their parents. Still, in the DIAN cohort, careful analysis of age of onset has shown that it is more predictable, within a narrow range of a few years, than previously thought for most mutations in a given family (Ryman et al., 2014).
GENFI’s suggested sequence of overall changes jibes with previous studies. The insula is a hub of the salience network that is known to be important in FTD, and subtle presymptomatic changes in neuropsychometry and brain imaging are known from isolated cases and smaller series (e.g., Jannssen et al., 2005).
In fact, at ICFTD Lize Jiskoot from Erasmus Medical Center in Rotterdam, Netherlands, presented follow-up data from such a study (Dopper et al., 2013). Liskoot said that two years after baseline, the 39 asymptomatic mutation carriers in her group had not improved on the neuropsych tests from one sitting to the next; unlike the non-carriers, the carriers had no practice effect and did not learn. On functional MRI, the carriers had a decline in the insula; across different imaging techniques used, they declined in frontal areas of the salience network and white-matter tracts connecting those areas, such as the uncinate fasciculus. Jiskoot’s site participates in GENFI.
Also in Vancouver, Emma Dowds of the University of British Columbia presented her group’s finding that eight presymptomatic carriers of a C9ORF72 repeat expansion had a thinner cortex than their unaffected relatives in the right insula and other frontotemporal regions; this was before their cognition declined. UBC is a site in the upcoming North American LEFFTDS cohort study (see Part 4 of this series). For their part, Suzee Lee and colleagues from UCSF showed reduced gray matter in a range of cortical and subcortical regions in C9ORF72 expansion carriers on their poster at ICFTD, but they noted that these differences might be developmental rather than degenerative. For a review of previous work in presymptomatic FTD, see Rohrer et al., 2013.
The next phase of GENFI will start in early 2015 as a five-year study of 600 participants. It will use largely the same protocol plus tau PET. Additional sites in Spain, France, and Germany have joined. To date, financial support for GENFI2 remains piecemeal, with sites scraping together local funding but no overarching grant approved yet.
GENFI2 aims to flesh out the emerging picture with much more data. The scientists want to address the twin questions of when during the presymptomatic decade might be the right time to intervene therapeutically, and how to measure whether the treatment works. To help plan future presymptomatic trials in FTD, Rohrer obtained EU funding for a working group called PreNI. It convenes experts from other neurodegenerative diseases who are conducting similar observational studies and have based therapy trials on them, such as TrackHD. PreNI includes U.S. advisers from DIAN. “We can learn lots from studies that have gone before us,” said Rohrer. The group will meet for the first time this month in London.
This EU-funded group works toward therapeutic prevention trials.
Perhaps the most difficult part of presymptomatic cohort studies is to find participants. Once a person has decided to enroll, they tend to be keen to enter a treatment trial, and many participants find purpose in contributing to research. In DIAN, which began in 2008, about 40 percent of the more than 400 participants have since decided to find out their genetic status, too, according to Randall Bateman of Washington University, St. Louis. But not everyone jumps at the chance. Just days before ICFTD, Rohrer said, he had met with a C9ORF72 family and explained GENFI to them, only to hear the day before his talk that they had politely declined to join. “They have grown up with FTD and know it is in the family. Still, they do not want to face it every year when they come to the visit,” said Rohrer. Working with carriers is new territory for neurologists. Rather than seeing symptomatic patients who come to their clinic, they have to cultivate an atmosphere of trust with younger, still-healthy people who harbor fears of an uncertain future.
In Germany, Felix Mueller-Sarnowski, a neurologist at Ludwig-Maximilians-University Munich, crisscrosses the country by train paying home visits to far-flung families with suspected autosomal-dominant Alzheimer’s disease in an effort to build that trust for the German DIAN study. “In the process, I also meet families with FTD. We cannot expect those families to open up easily. We have to support them,” Mueller-Sarnowski told Alzforum. In the United Kingdom, GENFI has set up a local “arms-length” support group, where family members can meet and talk about their experiences with FTD and with GENFI without a GENFI physician listening in. Rohrer jointly edits the website FTD talk www.ftdtalk.org to inform patients and families about FTD research.
In many instances, presymptomatic carriers come to the attention of scientists when their symptomatic relative sees a specialist who notices an autosomal-dominant inheritance pattern in the family history and invites the relatives to learn more about research opportunities. While done locally, this is beginning to coalesce in a more organized way as cohort studies of symptomatic FTD are coordinating with presymptomatic studies nationally and even internationally. In the United States, a push for such twin studies got an infusion of federal funding (see Part 4 of this series). In Germany, clinicians studying a large symptomatic cohort are now reaching out to its autosomal-dominant member families about GENFI2 (see Part 3).—Gabrielle Strobel
German Network of 700 FTLD Patients Presents Baseline Data
An 11-center consortium of clinical centers in Germany has enrolled 700 patients with symptomatic frontotemporal lobar degeneration (FTLD), most at early stages of disease, plus 10 presymptomatic at-risk relatives. Coordinated by Markus Otto of the University of Ulm, and funded by the government, the Multicentric FTLD Consortium’s Study reported data from its baseline visit at the 9th International Conference on Frontotemporal Dementia, held October 23 to 25 in Vancouver, Canada. Participants are returning for their follow-up visits, and the investigators are optimistic that they have enough cooperation from patients and their families to turn the project into a long-term natural history observation of the progression of FTD.
Centers of the German Multicentric FTLD Consortium Study. [Image courtesy of Markus Otto.]
This network includes patients across the clinical range of FTLD subtypes, from behavioral to language, parkinsonian and ALS-like, as well as some who have a still-undefined neurologic disease. It also includes people with Alzheimer’s disease and amyotrophic lateral sclerosis (ALS) as positive controls of these two ends of the spectrum, as well as neurologically healthy people as negative controls. The standardized protocol includes a similar range of clinical, neuropsychological, brain imaging, and fluid biomarker assessments as the Genetic FTD Initiative (GENFI) (see Part 2 of this series), and it also calls in its participants once a year. This deep phenotyping and biomarker study aims to capture transitions between clinical syndromes as patients progress. Another goal is to define both the differential diagnosis and outcome markers for use in therapeutic trials.
The German FTLD consortium accepts both sporadic and familial cases, in hopes of using their DNA for research on additional risk or causative gene variants. Two-thirds of the participants thus far have been genotyped, and more have expressed interest, Otto told Alzforum. Both participants and study staff are blinded to genetic results; people who want to know their status undergo genetic counseling and separate clinical-grade testing.
At ICFTD, the researchers presented baseline data on some 500 FTLD patients. As in GENFI, the achievement at present lies largely in having established a cohort and executing a standardized protocol across centers. People with FTLD can be socially disinterested or apathetic, raising the question of whether they will consent to altruistic commitments such as donating their time and tissue samples to research.
“We report no sensational scientific news from baseline,” Otto said. “But we can confirm the data from small, single-center studies in our large multicenter cohort, and show that we cover the entire spectrum of FTLD. It’s a robust foundation for longitudinal observation.” One worry with longitudinal cohort studies that ask a lot of their participants tends to be: Will they come back? In this study, 200 patients thus far have completed their first follow-up visit, and compliance is good, Otto told Alzforum.
Some data snippets: CSF analysis of 280 patients suggests that tau and phospho-tau are elevated in the logopenic variant of PPA (which is thought to be due to Alzheimer’s pathology). Among patients with behavioral variant (bv)FTD, tau was up in tau mutation carriers but not C9ORF72 mutation carriers. One patient with FTD symptoms and frontal atrophy turned out to have a presenilin mutation; he had an AD-like CSF signature of elevated tau/reduced Aβ. Progranulin was down in progranulin mutation carriers. Differences to some prior reports came up with neurofilament, an emerging marker in the FTD spectrum (Landqvist Waldo et al., 2013; Scherling et al., 2014). In the German cohort, neurofilament levels were elevated only in those subtypes of FTLD that had an element of ALS in their symptoms, said Otto. None of the other clinical FTLD diagnoses showed significant changes in these markers, or in CSF Aβ.
On brain imaging of FTLD subgroups at baseline, volumetry of 19 patients with progressive supranuclear palsy (PSP) showed shrunken gray matter in the basal ganglia, brainstem, and cerebellum, whereas 20 people with corticobasal degeneration (CBD) had shrinkage in their left frontal cortex. Twenty participants with ALS-bvFTD had lost gray matter in both sides of their frontotemporal cortices, as well as subcortically in the striatum and hippocampus. Sixteen people with bvFTD had atrophy in the posterior insula and in other cortical and subcortical regions. Thirty people with PPA had atrophy in a wide range of cortical and subcortical regions corresponding to language networks. In toto, these studies validated the consortium’s criteria for image acquisition and processing. They came in with low variability despite using different scanners at the respective sites, the scientists claim.
Matthias Schroeter at the Max Planck Institute for Human Cognitive and Brain Sciences, Leipzig, a consortium site, used the study’s data to evaluate the imaging component of new diagnostic criteria for the FTLD subtypes (Rascovsky et al., 2011; Gorno-Tempini et al., 2011). Schroeter subjected the network’s multimodal data to a process called “quantitative anatomical likelihood estimate meta-analyses,” which adheres to the PRISMA statement on reporting standards in health care research. This type of meta-analysis, Schroeter said, will be able to pinpoint the neural correlate for FTLD subtypes. By using machine learning and pattern recognition, researchers one day will be able to use data from multimodal imaging of a given patient for an earlier-stage differential diagnosis than clinical methods.
Like GENFI, the German network started in 2011 (Otto et al., 2011). It receives funding from the Federal Ministry of Education and Research (BMBF) until 2017 but hopes to continue beyond that, said consortium member Janine Diehl-Schmid of the Technical University in Munich. Otto told Alzforum that the German consortium is open to harmonizing and sharing its data with international cohorts so that it can be pooled and boost scientific power for analysis.
Diehl-Schmid, along with Manuela Neumann of the University of Tuebingen, will co-host the next ICFTD conference in Munich in 2016.—Gabrielle Strobel
Meet the Artful Leftie: NIH Jump-Starts U.S.-Canadian FTLD Cohorts
On October 23, on the day the 9th International Conference on Frontotemporal Dementia got underway in Vancouver, Canada, the National Institutes of Health released news of three five-year grants totaling $5.9 million for longitudinal cohort studies and pathobiology research. While that may not seem like big money in some fields, in FTLD it is. “They are the largest grants the NIH has ever funded for frontotemporal dementia,” said Tony Phelps of the National Institute on Aging (NIA). The money is a welcome shot in the arm for U.S.-Canadian initiatives in presymptomatic and symptomatic FTLD, similar to such projects already underway in Europe (see Parts 2 and 3 of this series).
The NIA, with its sister organizations the National Institute of Neurologic Disorders and Stroke (NINDS) and the National Center for Advancing Translational Sciences (NCATS), put their heads and their wallets together to help separate, ongoing U.S.-Canadian FTD cohort studies coalesce in a larger way. The biggest slice, $3.4 million, goes to a U.S. version of the Genetic Frontotemporal Dementia Initiative (GENFI). The study is called the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS—scientists pronounce it “Lefties”). Co-funded by NIA and NINDS, this multicenter study will be run by Bradley Boeve at the Mayo Clinic in Rochester, Minnesota, with Howard Rosen of the University of California, San Francisco, as co-principal investigator. The rest of the money is split about evenly. Adam Boxer at UCSF will lead a multicenter initiative to phenotype and genotype a broad-based cohort of FTLD. His team will characterize disease progression over one year, and build a consortium for therapeutic trials in such “readiness cohorts.” NINDS and NCATS co-fund this grant at $1.25 million. Thirdly, Leonard Petrucelli received $1.3 million from NINDS to explore, in his and other laboratories at the Mayo Clinic in Jacksonville, Florida, how C9ORF72 hexanucleotide repeat expansions cause FTD and ALS (see Part 7 of this series).
The LEFFTDS project engages seven centers in the United States, plus the University of British Columbia in Vancouver. LEFFTDS foresees longitudinal observation of 300 people in families who have inherited a pathogenic mutation in progranulin, C9ORF72, or tau. One hundred patients will be symptomatic mutation carriers, 100 will be asymptomatic mutation carriers, and 100 asymptomatic mutation non-carriers. “With LEFFTDS we are now studying asymptomatic mutation carriers, and do so specific to their mutation. We have never done that before,” said David Knopman, also at Mayo in Rochester.
Longitudinal MRI scans of a person carrying a pathogenic mutation in the gene encoding the tau protein, who becomes symptomatic during this time. [Image courtesy of Bradley Boeve, Mayo Clinic.]
Each of the participating sites already follow autosomal-dominant cases of FTD; indeed, all envisioned LEFFTDS participants are known to the centers, though if more come forward they will be allowed in, Boeve told Alzforum. Between the centers, the researchers currently work with 835 people from 306 families. About 100 are asymptomatic mutation carriers; an additional 300 asymptomatic relatives remain to be genotyped. LEFFTDS will enroll people starting in their 30s and invite them to their respective center once a year; it is currently funded for three such visits.
This study will use clinical, neuropsychological, biofluid, and neuroimaging measures, though not tau PET at present. As is becoming apparent in GENFI, LEFFTDS researchers also expect that they may see patterns of sequential change in those markers that are specific to each causative gene. Unlike in autosomal-dominant Alzheimer’s, the three main FTD genes are not known to impinge on the same molecular pathway. Much like in TrackHD, the Dominantly Inherited Alzheimer Network (DIAN), and GENFI, the overarching twin goals of LEFFTDS are to understand how the underlying proteinopathy causes disease and to determine how future disease-modifying trials can measure whether a drug is effective and engaging its target.
The second grant is called Advancing Research and Treatment for Frontotemporal Lobar Degeneration, or ARTFL. (Unsure how to pronounce this new acronym? Try “Artful”.) Previously this was called the FTLD Clinical Research Consortium. The overall goal here is to promote more therapeutic trials. From its headquarters at UCSF, ARTFL is working with six advocacy groups: the Association for Frontotemporal Degeneration (AFTD), the Bluefield Project, the Tau Consortium, CurePSP, CBD Solutions, and the Alzheimer’s Drug Discovery Foundation (ADDF). ARTFL engages the LEFFTDS sites plus six more, for a total of 14 sites across the United States, Toronto, and Vancouver.
ARTFL is designed to funnel an initially broad range of about 1,500 participants with any disease across the FTLD spectrum into a registry that is open to the public. From there, ARTFL will select about 900 participants for thorough characterization to build what are increasingly referred to as “readiness” cohorts for therapy trials. This includes a baseline workup of clinical, neuropsychology, imaging, and fluid markers for selected populations in whom the scientists believe they can determine which underlying proteinopathy drives their clinical disease. Moreover, ARTFL envisions a project to follow disease progression closely for one year, the time future FTD efficacy trials are expected to last. This serves to define progression scales and outcome markers. Finally, ARTFL includes genotyping for gene discovery; those participants in whom an autosomal-dominant mutation crops up would be offered a spot in LEFFTDS and/or in therapy trials for gene-specific subtypes of FTLD, said Boxer.
With GENFI, LEFFTDS, ARTFL, and the German and other networks, there are now a number of FTD consortia across the Atlantic Ocean. GENFI2, LEFFTDS, and ARTFL each expect to start up in spring 2015 after spending the winter preparing sites and obtaining permission of institutional review boards, etc.
The projects’ leaders each profess interest in making the data from their respective initiatives compatible to pooled analysis. Beyond the obvious lure of gaining greater statistical power for research on a rare disease, coordination has other upsides. For example, by sharing demographic, clinical, and genetic data internationally, centers can build better pedigrees. “If we are following siblings and cousins in the U.S., and a branch in Europe turns out to descend from the same founder mutation, then we can base age of onset on more data,” Boeve told Alzforum. Knowing when a mutation carrier will become symptomatic is important to decide when to start treatment, and to determine if putative disease-modifying agents delay onset. To link participant data across continents in a secure way, Boxer suggested the different studies set up a global unique identifier system (GUID), a code that would work much like a social security number for FTD.
A global, well-characterized patient population would attract more companies to developing therapeutics for this indication. It might also enable FTD researchers to act as a unified group in negotiating with industry, the study leaders agreed. “We want to assure that we have good publications coming out of the trials, and that data and biospecimens will be shared so that we can advance the science,” Boxer said. Toward this goal, Jonathan Rohrer of University College London, who co-leads GENFI, suggested that the study leaders meet periodically to exchange what they learned. He offered an acronym—FPI, for FTD Prevention Initiative—and extended an invitation to meet in Europe in summer 2015.
In 2011, DIAN created a pharma consortium with more than a dozen member companies that jointly fund trial preparation research and meetings. The consortium companies nominate their drugs for clinical trials in the DIAN population, but an independent committee of clinician-researchers then makes the choice (Dec 2011 news story). In addition, the leaders of DIAN, the Alzheimer’s Prevention Initiative (Feb 2010 news story), and the A4 study founded a group called CAP (aka Collaboration to Prevent Alzheimer’s), which meets several times a year to coordinate assessments and procedures as closely as possible (Aug 2012 news story).
As another area of global collaboration, Boxer suggested a jointly funded global patient-research registry. This might work well across international borders if it was built as a federated platform where certain core information could be merged but other components are maintained locally in separate, national registries, Boxer said at ICFTD. Prior examples include the Global Dystonia Registry, the Brain Health Registry started at UCSF, and the Alzheimer’s Prevention Registry.
Goodwill notwithstanding, there are limits to cross-Atlantic cooperation. For example, in its effort to nudge the North American studies to collaborate and contribute to existing data platforms in Alzheimer’s disease, the NIH likes to see its funded projects deposit de-identified data in databases such as the database of Genotypes and Phenotypes (dbGaP). ARFTFL and LEFFTDs will also upload their imaging data to the place where ADNI data lives, the Laboratory of Neuro Imaging (LONI) at the University of Southern California. Moreover, both initiatives will use the NACC-FTLD test module. This was created as an outgrowth of the Unified Data Set collected as part of the U.S. Alzheimer Disease Research Centers system, and the tests are available for free without licensing fees (May 2012 news story). For its part, GENFI uses an overlapping but not identical set of tests and translates them into many languages. The NIH also requires that ARTFL and LEFFTDS contribute blood and other biospecimens to the National Cell Repository for Alzheimer’s Disease (NCRAD) and the NINDS Coriell Repository, which collect information from NIH-funded studies for use in neurogenetics and other research.
ARTFL and LEFFTDS nabbed their new funding in part because they had been laying the groundwork in smaller projects. On those, the ICFTD featured various data updates. For example, the Neuroimaging in Frontotemporal Dementia project is an imaging study in symptomatic FTD. The NIFD is in the midst of collecting second scans on 74 people with bvFTD, 34 with svPPA, 33 with nonfluent PPA, and 73 controls. In his talk, Rosen of UCSF said that he expects the regions that atrophy in a given patient will predict which symptoms that person develops later, and that the study can define rates of atrophy in specific brain areas against which to measure future drug effects.
This image shows where MRI scans taken one year apart in two types of FTLD measured the greatest loss of brain tissue. Red-yellow areas indicate the relative severity of atrophy in people with behavioral variant frontotemporal dementia (bvFTD), whereas yellow-orange-blue areas show annual tissue loss in progressive supranuclear palsy (PSP). Color images superimposed on MRI of an healthy adult brain. [Image courtesy of Shubir Dutt, Howard Rosen, Adam Boxer, UCSF.]
Already, short stretches of one-year longitudinal data suggest that atrophy in defined areas starts presymptomatically, accelerates when symptoms set in, and bottoms out in the overt symptomatic phase, Rosen said. In essence, the FTLD field is beginning to validate theoretical biomarker staging diagrams with empirical data from longitudinal observation, as Alzheimer’s research has been doing for some years. With the new grants, several of these studies can now band together for a better-funded continuation of their ongoing work.
Rosen compares his site’s data with those of Brad Dickerson at Massachusetts General Hospital, to see if the two sites come up with similar results. “We are very close,” Rosen said in Vancouver. The combined data serve to plan future treatment trials. For example, analyzing reams of existing scans across the two sites has led to a realization that FDG-PET—an established technique that infers brain activity in a given region via that region’s glucose use—may not be worth the expense in large FTD trials, Rosen claimed. Brain maps made by various MRI techniques, such as arterial spin labeling, are proving as good. “There may not be added value of FDG PET over MRI,” Rosen said.—Gabrielle Strobel
Too Hot, Too Cold, or Just Wrong? Physiology Links Behavior to Circuits in FTD
Walking around in public without clothes, or baking in the summer sun with much too much on? Laughing at a funeral, or frowning at a funny joke? These are just a few of the odd behaviors that the partners of people with frontotemporal dementia (FTD) first notice about their loved one. Sometimes the abnormalities seem to contradict each other—some people with FTD become hypersensitive to certain stimuli, while others grow a thick skin. How are these diverse symptoms related, and what are the underlying changes in neural circuitry that cause them? How can doctors reliably detect early changes in something as varied as behavior? Could these changes be clues to diagnostic or therapeutic biomarkers? Researchers grappled with these questions, and presented some early answers, at the 9th International Conference on Frontotemporal Dementias, held October 23 to 25 in Vancouver, Canada.
Whatever their nature, FTD-related behavioral changes all represent a deficit in the brain’s ability to process, interpret, or respond to incoming stimuli such as pain, temperature, sound, emotion, or social cues, said Virginia Sturm of the University of California, San Francisco. Sturm and other researchers presented clever ways to measure patients’ responses to these stimuli via physiologic and autonomic responses, such as dilated pupils, a quickening of the pulse, or a spike in blood pressure. They then correlated those findings with neuroimaging and genetics data to uncover the networks that break down in FTD. One common vein in their findings was the disintegration of hubs in the cortex's salience network—such as the insula and the cingulate—in people with FTD who lose their ability to respond properly to emotional or physiologic stimuli. People with C9ORF72 mutations, a major genetic cause of FTD, had trouble even deeper in the brain, in the thalamus. From this coupling of physiological phenotypes with neurodegenerative patterns, key “FTD circuits” started to materialize at the conference.
While we think of frontotemporal dementia as a disease of behavior, executive function, and cognition, it also has a sensory component. Perceptions and interpretations of heat, pain, even body image, can all be impaired early on.
[Image courtesy of Thomas Haslwanter.]
Jason Warren of University College London broke the ice on this topic during the first session of ICFTD. Warren presented data on pupil changes in response to sound. He compared responses of patients across the spectrum of FTD disorders, as well as people with Alzheimer’s disease and healthy controls. In general, people with FTD responded less robustly to sounds than did the two control groups. However, key differences even between different types of FTD emerged when Warren compared the way people responded to a meaningless sound, such as static noise, to the way their pupils ballooned when hearing a sound with real-world meaning, such as the buzz of a mosquito.
Unexpectedly, people with semantic dementia had the biggest difference in their responses to meaningful versus meaningless sounds. They were aroused by meaningful sounds, even though their semantic deficits prevented them from understanding what those sounds meant. Warren thinks the results suggest that the brain may be hyperstimulated by salient sounds it cannot define. “This could be an autonomic index of things not being quite right with the world, similar to when you hear something that goes bump in the night,” he told Alzforum. Interestingly, this test held up as a way to measure semantic deficits across all the disorders. For example, people with AD who displayed semantic deficits on other tests also tended to have a larger pupil response to salient sounds. This test therefore could be useful as a physiological measure of semantic decline, or perhaps in the setting of therapeutic trials of recovery of semantic memory.
Warren’s group presented a slew of posters on the first day of the conference, displaying data on the way people with FTD responded to music, humor, art, and even sarcasm, all of which were off in some manner. To connect these response deficits to underlying neural circuitry, the London group analyzed the way people with FTD responded to pain and temperature changes. Phillip Fletcher, a graduate student in Warren’s lab, presented these findings in a talk.
Many people with FTD show early abnormalities in their ability to feel or process pain or changes in temperature. To get an idea of how these symptoms present across the FTD spectrum, Fletcher gave caregivers a questionnaire that asked for details about their loved ones’ responses to temperature and pain. Fletcher found that 32 out of 58 patients with FTD had problems with pain and/or temperature awareness, but the problems manifested in different ways between clinical types. People with semantic dementia tended to have exaggerated responses, often complaining of cold or pain, for example. On the other hand, people with behavioral variant FTD tended to have dulled responses. One caregiver recalled having to insist that a patient remove his jacket when it was hot outside; another reported that a patient did not complain of pain even after suffering a bad fall and a black eye. All six patients with C9ORF72 mutations (five with behavioral variant FTD, one with progressive aphasia) had abnormal pain and/or temperature awareness.
To correlate these abnormalities with neuroanatomical changes, Fletcher and colleagues scanned the patients for gray-matter atrophy using voxel-based morphometry. All patients with disturbed temperature or pain awareness had atrophy in their right posterior insula. This makes sense, Fletcher said, because the region has been shown to switch on in response to pain. Patients lacking C9ORF72 mutations also had shrinkage in the right anterior temporal lobe, a region that is important for nonverbal semantic processing. In contrast, the C9ORF72 expansion carriers had normal anterior temporal lobes but a shrunken thalamus—one of the first regions to encounter and process incoming pain and temperature signals. From there, the signals move up toward the posterior insula, which creates a virtual map of the signal’s origin in the body. The posterior insula then communicates with the anterior insula, which integrates this sensory information with signals from the cortex, helping put the pain in context. Together, these regions coordinate the pain or temperature response.
That C9ORF72 mutation carriers have atrophy in the thalamus jibes with other studies. Laura Downey, also from the University College London group, recently found that C9ORF72 mutation carriers have skewed body schema, meaning they have difficulty creating a mental representation of their body or determining where their own body ends and another’s begins (see Downey et al., 2014). Body schema mapping relies heavily upon both the insula and the thalamus. Another recent study led by William Seeley at the University of California, San Francisco, found that C9ORF72 carriers had more thalamic atrophy than patients with other types of FTD, and that atrophy correlated with decreased functional connectivity between the thalamus and key hubs of the salience network, including the anterior insula and cingulate cortex (see Lee et al., 2014).
FTLD has long been considered primarily a cortical disease, and the role of the thalamus has not been appreciated until recently, Warren said. “The thalamus has been forgotten and then ‘rediscovered’ throughout the history of neurology,” he said. The recent combination of physiological and neuroimaging data has built a strong case for the region’s importance in particular dementia diseases. “The physiological markers and the anatomy data are all gelling,” he said.
Downey and Colin Mahoney, another graduate student from Warren’s group at UCL, delved into the changes in structural connectivity that underlie some FTD patients’ difficulties in picking up social cues, such as sarcasm. Warren asked patients to interpret the intent of actors in a video to determine their grasp of sarcasm or other emotional states. Then, the researchers performed diffusion tensor imaging to measure the integrity of white-matter tracts. A defunct “sarcasm radar” correlated with decreased integrity of the uncinate fasciculus, a white-matter tract that connects regions of the limbic system to those in the cortex. A patient’s inability to read emotions in others correlated with an erosion of white-matter tracts emanating from the thalamus and the fornix.
In her talk, Sturm dug deeper into complex emotions and social cues. People with FTD often lack inhibition or a sense of decorum, and Sturm attempted to take stock of this deficit using an amusing test. She presented data from a study where researchers measured embarrassment in FTD patients with the “karaoke task.” Participants were fitted with headphones and asked to sing along to the song “My Girl” while being video-taped. They then had to watch themselves singing the song without the accompanying music—an experience that would make most people cringe. During the playback, the participants were monitored for signs of embarrassment, such as changes in facial expression, as well as autonomic responses, such as increased heart rate, sweatiness, or blood pressure. As expected, people with behavioral variant FTD had fewer changes in facial expression or autonomic responses while watching themselves than other people did (see Sturm et al., 2013). Neuroimaging data revealed that this blunted self-consciousness correlated with atrophy in the anterior cingulate cortex, another key hub in the salience network.
It makes sense that the anterior cingulate cortex (ACC) would be involved in generating feelings of embarrassment, Sturm said, because it has connections with the frontal lobes, which facilitate the understanding and encoding of social context and roles, as well as with regions deeper in the brain that mediate autonomic changes in heart rate and breathing. “The ACC acts as a way station between understanding a social context and mounting an emotional response to it,” Sturm said. “So you could imagine that complex emotions, such as self-consciousness, might be particularly hard hit if this hub is lost.”
Sturm is applying similar methods—measuring facial expressions and autonomic responses—to probe empathy in FTD. She monitored patients’ faces while they watched a heart-warming scene from a movie, looking for signs that the patients felt the same way. People with AD had heightened empathic responses compared to controls, while the scene left people with FTD cold. Sturm is currently searching for neuroimaging correlates of this lack of empathy. Similar regions may crop up, as previous studies have reported that the posterior insula lights up when someone experiences pain, and the anterior insula and ACC are activated when someone watches a loved one experience pain (see Singer et al., 2004).
Linking these physiological measures with changes in brain structure finally anchors emotion in biology, Sturm said. “Many times people don’t think emotion is biological; they think it’s something else,” she said. “This work is important for pinpointing the biological reason in the brain why people change their emotions and behavior, and for differentiating it from just a psychiatric explanation.” She added that such studies will also inform researchers’ understanding about the networks that may be affected in psychiatric disorders.
One major theme at ICFTD was that FTD is a behavioral disease. “In the past, we’ve stressed executive function, but now there is more of a push to look at the social/behavioral context of FTD,” said Nadine Tatton of the Association for Frontotemporal Degeneration (AFTD). While physiological measures have a long way to go before they will be seen as objective, standardized tests employed in many clinics, they could ultimately make behavioral symptoms more tractable, Tatton said.
These symptoms initially may be subtle. Work led by Camilla Clark in the UCL group suggests that changes in a person’s sense of humor may be an early warning sign of FTD. Warren views physiological measurements as a way to offer better explanations to patients and caregivers. For example, a person may be perplexed about why their father suddenly switches from enjoying Woody Allen movies to getting a kick out of the Three Stooges. “When it comes to these more complex behavioral and emotional changes, we’re really not giving people answers, and I’d like to change that,” he said. “Even before we have a treatment, I’d like to give people a useful consultation.”
Warren believes distilling emotional responses into relatively simple tests and measurements will entice neurologists to follow suit. “Clinical neurologists are starting now to genuinely engage with the need to look at things like social cognition and emotion, something psychiatrists are more used to dealing with,” he said.
Warren hopes that the measures may one day aid diagnosis or help monitor progress in clinical trials. “The main point of measuring physiological markers is to deconstruct things that are relatively complex into things that are reproducible, measureable, and may appear prior to structural changes in the brain. Tests that seem off-the-wall, like the karaoke test, might be what we need to know whether an intervention is working or not,” he said. “Otherwise, how else would you measure?”
Dana Hilt of FORUM Pharmaceuticals in Watertown, Massachusetts said that some form of physiological test could serve as a bridge between biochemical markers, such as blood biomarkers or neuroimaging, and clinical outcomes, such as overt changes in behavior and executive function. Hilt used such a measure—evoked-response EEG measurements—in a schizophrenia trial to learn if the drug not only accessed the CNS, but triggered a pharmacological effect (see Preskorn et al., 2014).
“What I think is necessary in this area are new ‘intermediate biomarkers’ that are stronger than biochemical effects and that can then be used to select doses and schedules for full clinical studies,” Hilt wrote in an email to Alzforum. “Assessment of cognition and clinical function are impacted by so many sources of variability that it makes utilizing these for smaller Phase 2 studies quite risky. An ‘intermediate biomarker’ would be useful in this regard.”—Jessica Shugart
Progranulin-Boosting Drug Moves into Phase 2 for Frontotemporal Dementia
When it comes to clinical trials, frontotemporal dementia has long gotten the shaft. The only currently approved FTD drugs were first tested in other disorders, and they aim to soothe symptoms rather than attack the cause of the disease. Most FTD trials, for example one of memantine, in fact were negative (see Boxer et al., 2013). A new Phase 2 study now offers hope that this could change. At the 9th International Conference on Frontotemporal Dementias, held October 23 to 25 in Vancouver, Canada, researchers from FORUM Pharmaceuticals in Watertown, Massachusetts, announced a step forward in testing FRM-0334, a small-molecule inhibitor of histone deacetylase (HDAC). Researchers hope the compound will boost the expression of progranulin, a neurotrophic factor that takes a hit in those FTD patients who harbor pathogenic mutations in the granulin gene.
The next FRM-0334 trial will only target the roughly 20 percent of familial FTD patients who harbor mutations in the gene for progranulin. Even so, researchers expressed excitement that a bona fide FTD trial was moving forward. “Clinical trials are where the rubber meets the road,” said conference host Howard Feldman of the University of British Columbia in Vancouver. Researchers have come to agree that gene-based drug discovery makes sense given that the FTLD group of diseases has proven to be both genetically and clinically heterogeneous. “We need to use the tools that are in front of us, and hopefully any knowledge we gain with the genetic forms of the disease will inform the sporadic disease as well,” said Susan Dickinson, executive director of the Association for Frontotemporal Degeneration (AFTD). FORUM’s Holger Patzke presented preclinical findings and laid out plans for the upcoming trial. “We are really excited to put a stake in the ground,” he told Alzforum.
Researchers discovered in 2006 that mutations in the granulin gene caused FTD, shortly before finding that TAR DNA binding protein-43 (TDP-43) dominated the inclusions found in the same patients. Since then, 70 FTD-linked progranulin mutations have been identified. All are predicted to reduce progranulin production, and mutation carriers have lower serum levels of the progranulin protein (see Jun 2009 conference story). Full-length progranulin is shed from cells and processed into several smaller granulins, which have been reported to promote neural growth and regulate inflammatory responses (see Oct 2012 news story). Researchers don’t know how progranulin deficiency hastens FTD, but the fact that its reduction causes the disease makes progranulin a particularly tantalizing target for therapy, Patzke said.
Researchers have proposed different strategies to boost progranulin levels, including blocking sortilin, a progranulin receptor that removes the protein from circulation (see Aug 2012 news story). Some progress on that front in animal studies was presented at ICFTD, but FORUM’s approach is by far the furthest along.
FORUM scientists decided to look for HDAC inhibitors that would boost transcription of the non-mutated copy of the gene. HDACs work by deacetylating chromatin, which keeps genes closed off from transcription. Previously, researchers led by Joachim Herz at the University of Texas Southwestern Medical Center in Dallas had identified suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved by the FDA to treat T-cell lymphoma, as boosting progranulin expression (see Cenik et al., 2011).
FORUM had been working for more than a decade to develop HDAC inhibitors that would penetrate the central nervous system. They eventually selected FRM-0334, a compound that induces histone acetylation in the rodent brain at 1/10 the dose of SAHA. At ICFTD, Patzke reported that the compound boosted progranulin mRNA and protein levels in rodent neurons and microglia. In lymphoblasts derived from FTD patients with progranulin mutations, it raised progranulin protein to near normal levels. A single oral dose of the compound efficiently crossed the mouse blood-brain barrier and boosted progranulin levels in the cortex.
Previous studies on other HDAC inhibitors had already shown promise, said Nadine Tatton, scientific director at AFTD. “Now, to see something that is more refined and effective at a nanomolar scale is exciting.”
Patzke summarized results from FORUM’s recently completed Phase 1 trial to test safety and tolerability of the compound in healthy people. Seventy volunteers received daily oral doses of FRM-0334, escalating to 400 mg over two weeks. The researchers observed no severe adverse events. The trial did not reach what is called the maximum tolerated dose, the highest amount of drug that does not cause unacceptable side effects, and the scientists concluded the treatment was well-tolerated.
To move on to Phase 2, the researchers needed to make sure that they would be able to accurately measure levels of progranulin in the CSF and blood plasma of patients. If levels fluctuated much between participants, or worse, within a given participant at repeat measures, then interpretation of the drug’s potential effects could become difficult. The goal of the trial is to restore normal progranulin expression in mutation carriers, which gives researchers no more than a twofold window of wiggle room. As presented by FORUM’s Gordon Loewen on a poster at the conference, the researchers evaluated performance of an ELISA kit and tracked CSF progranulin levels in healthy volunteers as well as in people with Alzheimer’s disease or mild cognitive impairment. Over two different sampling periods spaced two weeks apart, progranulin levels varied by only 8.6 percent within subjects. In collaboration with John van Swieten’s lab at Erasmus Medical Center in Rotterdam, Netherlands, plasma progranulin levels turned out to remain remarkably steady in carriers of granulin mutations as well as non-carrier controls. “It is actually stunning how stable progranulin levels are,” Patzke said during his talk.
The Phase 2 clinical trial, which will be double-blind, randomized, and placebo-controlled, is set to begin in 2014, Patzke said. Thirty granulin mutation carriers in the symptomatic or prodromal stages of FTD will be enrolled at 14 sitesin the United States and Europe. The participants will receive one daily dose of FRM-0334, or placebo, by mouth for 28 days. In addition to safety and tolerability, the researchers will measure plasma and CSF levels of FRM-0334 as well as of progranulin (see clinicaltrials.gov) to measure drug exposure and target engagement. Patzke said he expects the trial to be complete by mid-2016.
In a separate effort, scientists at UCSF are currently evaluating nimodipinein eight granulin mutation carriers for its ability to increase levels of progranulin. Nimodipine is an FDA-approved calcium channel blocker that is being prescribed in acute care to prevent stroke in people who have suffered a subarachnoid hemorrhage.
While attacking FTD at its source is the ultimate goal, symptomatic treatments that assuage FTD’s behavioral symptoms are also needed. Caregivers can get overwhelmed dealing with loved ones who become cold or apathetic. On that score, Elizabeth Finger of the University of Western Ontario, Canada, ended the conference on a glimmer of hope. She treated a small number of behavioral variant FTD patients with escalating doses of intranasal oxytocin, a hormone known to promote nurturing (see clinicaltrials.gov). An abstract of Finger’s talk is published as part of the conference proceedings. Finger’s study has been placed under embargo by a journal until January, and Alzforum will report details at that time.
“For years, we’ve had no trials, which is a devastating message to give to patients and caregivers,” Dickinson said. “At the meeting, I heard researchers voice excitement that we are near effective therapies. I’ve never heard people speak of that in the same way before.”—Jessica Shugart
Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
When it first broke, the news that expanded sequences in the C9ORF72 gene are a major cause of both frontotemporal dementia and amyotrophic lateral sclerosis shook up both research communities (see Sep 2011 news story and Sep 2012 conference coverage). Three years later, research on this mutation has exploded, and was on full display at the 9th International Conference on Frontotemporal Dementias held October 23 to 25 in Vancouver, Canada. Researchers have spent this time generating tools to study the problem and are now ready to grapple with the questions that preoccupy them at this stage: How do the repeats wreak havoc on the nervous system? And why do some people with the expansions develop FTD, others ALS, and still others a combination of the two?
Tucked into an intron in the C9ORF72 gene, the simple sequence GGGGCC is harmless in small doses. Normal people have up to a couple of dozen repeats of the sequence, but people with ALS or FTD can harbor hundreds to thousands of sequential copies. Researchers estimate the run-on repeat accounts for 40 percent of familial ALS and about a quarter of familial FTD, as well as a smaller fraction of sporadic disease (see Majounie et al., 2012).
Once transcribed, the repeat sequences form secondary RNA structures called G-quadruplexes, which bundle up with RNA-binding proteins in nuclear aggregates called RNA foci. The repeats are intronic, yet they are translated in both the sense and antisense directions by a process called repeat-associated non-ATG, or RAN, translation. Depending on where in the mRNA translation starts and in what direction it moves, five different peptides can be produced, each made up of strings of just two amino acids: glycine-alanine (GA), glycine-proline (GP), glycine-arginine (GR), proline-alanine (PA), or proline-arginine (PR). These “dipeptide repeats,” or DPRs, have been reported to form intraneuronal inclusions (see Feb 2013 news story and Feb 2013 news story).
At the meeting, researchers proposed three possible ways in which C9ORF72 expansions might spell trouble for neurons. The sequestration of C9ORF72 RNA into foci has been reported to prevent its expression, although researchers do not yet know what C9ORF72 does. The other two possibilities revolve around a gain-of-function premise. In one scenario, the RNA foci muck up the expression of a plethora of genes by sequestering and withholding the services of crucial RNA-binding proteins, which play key roles in everything from RNA splicing to transport to translation. In the other, the expansion exerts its toxicity at the protein level: DPRs somehow damage neurons, either through the inclusions they form or, more provocatively, through soluble forms that researchers have yet to detect.
Where Do DPRs Fit In?
When severity is plotted against brain regions, TDP-43 pathology (blue) appears nearly in lockstep with neurodegeneration (red) in people with ALS (top panel) or FTD (bottom). In contrast, dipeptide repeats (green) do not line up with neuronal damage. [Image courtesy of Mackenzie et al., Acta Neuropathologica, 2013.]
Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida made a case implicating the DPRs. In co-discovering that RAN translation could beget DPRs, Petrucelli’s lab developed a C9-RANT antibody against the poly-GP DPR. The antibody revealed the existence of both cytoplasmic and nuclear inclusions in the brains of people with FTD/ALS who carried the C9ORF72 expansions. Unlike the repeat-laden RNA foci the researchers detected in cells throughout the carriers’ bodies, for the most part the DPR inclusions were only present in neurons in the brain (see Ash et al., 2013). Petrucelli interpreted those initial findings as an indication that the RNA foci do not closely correlate with DPR pathology. He hypothesized that transcript levels of the repeats dictated the development of inclusions.
In collaboration with Matthew Disney’s lab at the Scripps Research Institute in Jupiter, Florida, Petrucelli’s lab has since developed small-molecule inhibitors that bind to the RNA repeats and block the formation of both RNA foci and DPRs. They also developed an antibody assay that could detect GP DPRs in the cerebrospinal fluid (CSF) of C9ORF72 expansion carriers with ALS (see Aug 2014 news story). At the meeting, Petrucelli presented new progress on both fronts. He reported that his lab had developed stable cell lines to facilitate additional drug screening of compounds, a process that has so far quadrupled potency, Petrucelli told the crowd.
He has also expanded his CSF biomarker studies to include FTD patients with C9ORF72 expansions through collaborations with other institutions, including the University of California, San Francisco. His early findings include detection of soluble GP DPRs in the CSF of FTD patients, as well as asymptomatic C9ORF72 expansion carriers. GP concentrations rose in some patients from one year to the next, which could hint that DPRs are associated with progressive pathology.
During the ICFTD, the NIH announced it had given a P01 grant to Petrucelli and his Mayo Clinic colleagues to advance C9FTD/ALS research, which will award the researchers $1.3 million during the first year (see Part 4 of this series).
Adrian Isaacs of University College London reviewed recently published data implicating the DPRs in neurodegeneration in a Drosophila model. He had reported that the DPRs were both necessary and sufficient to cause C9ORF72-mediated neurodegeneration in the fly eye; RNA foci alone were not enough (see Aug 2014 news story).
A fly in this ointment came from Chris Shaw of Kings College London, who presented data that seemed to question the role of C9ORF72 DPR inclusions in neurodegeneration. Shaw first recapped results from a paper last year, in which his group had reported that RNA foci sequestered a number of key RNA-binding proteins and killed neurons in culture, whereas the cells that managed to translate DPRs fared better (see Lee et al., 2013). He also presented some data in which some DPRs, when expressed at high enough levels, could cause toxicity in cell culture.
However, Shaw said the connection between DPR inclusions and disease started to crumble when researchers compared the distribution of inclusions to that of neuropathology in the diseased human brain. To make this point, Shaw drew on dramatic data from a study led by Manuela Neumann of the University of Tübingen, Germany. In that study, which Neumann referenced in her talk at the conference, researchers compared the distribution of C9ORF72 GA DPR inclusions to that of TAR-DNA-binding protein 43 (TDP-43) inclusions and to neurodegeneration in C9ORF72 carriers with FTD, FTD/ALS, or ALS. As had been reported previously, they found that TDP-43 built up in cytoplasmic inclusions in people with these disorders, regardless of whether they harbored C9ORF72 expansions. The TDP-43 inclusions are thought to be toxic to cells.
Importantly, Neumann found that where there were TDP-43 inclusions, there was also neurodegeneration. Both were highest in the motor cortex and spinal cord in ALS patients, and in the frontal cortex in FTD patients. However, the distribution of C9ORF72 GA dipeptide repeat inclusions correlated hardly at all with neurodegeneration. It was similar between patients with FTD or ALS, with levels highest in the cerebellum, frontal and occipital cortices (see Mackenzie et al., 2013). Shaw went on to present new data revealing via western blot that the amount of insoluble TDP-43 inclusions was largely the same in FTD patients with or without C9ORF72 expansions, further disentangling C9ORF72 DPRs (at least GA) from TDP-43 pathology.
Although the role of TDP-43 in neurodegeneration is still not clear, the fact that it seems entwined with neuronal death indicates that it may arise late in the disease process. In support of this idea, Petrucelli mentioned a study led by Mackenzie in which a single C9ORF72 carrier died of a pulmonary embolism at the age of 26. He had childhood learning problems and became psychotic prior to his death, but had not been diagnosed with FTD. At autopsy, his brain was riddled with C9ORF72 GA DPRs, but not yet with TDP-43 inclusions (see Proudfoot et al., 2014). How C9ORF72 expansions are related to TDP-43 pathology—and why that TDP-43 pathology also occurs in patients who do not have C9 expansions—remains a mystery. Some researchers even suggested that the presence of DPRs could be a sign of a protective, rather than destructive, mechanism in certain neurons, for example in the cerebellum.
How should researchers proceed to uncover the role of the C9ORF72 in FTD/ALS? For starters, they would be well advised to compare the distribution of all known DPRs, not just focus on one or another, commented Ian Mackenzie of the University of British Columbia in Vancouver. Mackenzie co-authored the C9 distribution study with Neumann. He said that it will be important to determine whether soluble forms of the DPRs, rather than the more easily visible neuronal inclusions, play a role in neurodegeneration.
RNA foci are more widely distributed than either DPRs or TDP-43 inclusions. Mackenzie added that perhaps a key factor to consider is which cell populations are most susceptible. For example, although RNA foci can be found throughout the body, perhaps only some neuronal populations are sensitive to the sequestration of certain RNA binding proteins, he said. This selective vulnerability could dictate where in the brain neurodegeneration strikes.
Some of these questions might get answers with new mouse models presented at the conference. Clotilde Lagier-Tourenne of the University of California, San Diego, presented initial findings from transgenic mice expressing either 110 or 450 C9ORF72 repeats. She found RNA foci and DPRs only in the mice expressing 450 repeats. Boosting their expression level slightly enhanced RNA foci and DPR expression. Lagier-Tourenne plans to use these mice to study the relationship between C9ORF72 expansions and neurodegeneration, and to test whether targeting the repeats with antisense oligonucleotide (ASO) therapies can prevent it.
ASOs are RNA-like chemicals that are modified at the backbone to make them bind more potently to their target mRNAs. At ICFTD, ASOs got praise as an innovative way forward. Tim Miller of Washington University, St. Louis, presented data showing that targeting normal tau in this way to reduce tau protein levels improved a list of outcome markers in mouse tauopathy models from pathology to neuron loss, astrogliosis, and even seeding. An ASO therapy against SOD1 performed well on safety and pharmacokinetics in a Phase 1 trial for amyotrophic lateral sclerosis (ALS) (see Miller et al., 2013). Plans to develop ASOs against C9ORF72 are only just beginning, said Mackenzie, but the concept makes great sense. With C9ORF72, scientists could target only the pathogenic expansion. “If the expansion exerts a toxic gain of function, which most of us think, then you can prevent that,” said Mackenzie.
Reporting from the mouse front, Adam Walker of the University of Pennsylvania presented striking findings from a new TDP-43 transgenic line. These animals express a doxycycline-controllable TDP-43 gene that lacks its nuclear localization signal, relegating the protein to the cytoplasm. The mice rapidly deposited phospho-TDP-43 inclusions throughout the brain and spinal cord shortly after researchers switched on the gene. Severe motor-neuron disease followed soon, and the mice died by three months of age. The researchers can rescue motor deficits by switching off the toxic gene. Walker said he will use the mice to understand how TDP-43 pathology causes neurodegeneration, and to test out new therapeutics. One can’t help but wonder what might happen if Walker and Lagier-Tourenne’s mice got together.—Jessica Shugart
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, , Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ.
Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study.
Lancet Neurol. 2012 Apr;11(4):323-30.
PubMed.
For every other person with frontotemporal dementia (FTD), some form of the disease runs in the family. A few causative genes have already taken the blame for plaguing nearly half of these families, but much of the genetic burden remains unaccounted for. At the 9th International Conference on Frontotemporal Dementia, held last month in Vancouver, Canada, the ongoing hunt for more genetic culprits was on full display. Besides offering up a handful of new, potentially causal mutations for discussion, researchers presented a trove of data revealing how some genes steer the course of the disease in people who harbor a known variant. Another researcher probed the mechanistic underpinnings of a previously discovered mutation, and uncovered a fundamental finding about the way RNA moves throughout the cell.
Autosomal-dominant mutations in the genes encoding tau (MAPT), progranulin (GRN), and C9ORF72 account for the lion’s share of genetically inherited FTD cases in which a gene has been found. Mutations in the genes encoding valosin-containing protein (VCP), fused in sarcoma (FUS), and charged multivesicular body protein 2B (CHMP2B) are much rarer. Together, these known causal genes explain the disease in roughly 40 percent of affected families, plus about 10 percent among cohorts of unrelated patients. Complicating matters, even people who share the same causal mutation may present with drastically different disease characteristics. For example, people harboring C9ORF72 expansions may develop amyotrophic lateral sclerosis (ALS), FTD, or a devastating combination of both disorders. The age at which disease strikes can vary by decades. This bewildering picture has spurred researchers to push for progress in two directions: new genes that cause disease, and genetic factors that explain the range of clinical phenotypes.
The Modifiers Rosa Rademakers of the Mayo Clinic in Jacksonville, Florida, kicked things off with factors that influence disease in carriers of the C9ORF72 expansion. Rademakers touched on recently published data suggesting that the two genes TMEM106B and ataxin-2 modify which clinical disease develops in a given C9ORF72 carrier. Specifically, TMEM106B mutations protect C9ORF72 expansion carriers from developing FTD, but not ALS; similarly, an intermediate number of expansions in the ataxin-2 gene skewed C9ORF72 carriers toward ALS (see van Blitterswijk et al., 2014, and Sep 2014 news story).
To dig for more genes that may influence when a C9 carrier becomes symptomatic, Rademakers and dozens of colleagues screened some 300 C9ORF72 expansion carriers for 36 different genetic variants that previously had been associated with FTD or ALS. This netted three variants associated with when disease struck, plus six associated with the length of survival thereafter (see van Blitterswijk et al., 2014). The candidate modifier genes include those involved in protein sorting (ubiquitin-associated protein 1), transcription (elongator acetyltransferase complex subunit 3), and antioxidant defense (metallothionein), as well as APOE4 and granulin. All need confirmation in larger cohorts, Rademakers said.
Rademakers also presented fresh data looking at how C9ORF72 expansions trigger changes in gene expression in the brain. Performing whole-genome expression analyses on samples from 32 expansion carriers and 30 controls, her group identified 40 genes that were differentially expressed in the cerebellum, and three in the frontal cortex. A majority hailed from the homeobox, or hox, family of genes, which is primarily implicated in development but also in neuronal repair and regeneration. In preliminary experiments in non-neuronal cell lines, Rademakers found that loss of C9ORF72 expression (as opposed to expression of C9ORF72 expansions), triggered hox gene upregulation. The C9ORF72 expansion is known to generate gobs of dipeptide repeat (DPR) inclusions in the cerebellum but, oddly, this region is not particularly hard hit with neurodegeneration, Rademakers said (see also Part 7 of this series). The expansions also downregulate expression of the C9ORF72 gene itself. Rademakers speculated that the hox response could point to a protective mechanism that counters negative effects of the C9 expansion in the cerebellum.
In a packed talk, Christine Van Broeckhoven of the VIB, University of Antwerp, Belgium, laid out more findings about genetic modifiers. For one, Van Broeckhoven looked in C9ORF72 expansion carriers for evidence of genetic anticipation. In this phenomenon, known from Huntington’s disease, children develop the disease at a younger age than their parents did. Drawing on blood and brain samples from C9ORF72 family pedigrees, Ilse Gijselinck, a postdoc in Van Broeckhoven's group, found that expansions grew longer from one generation to the next, and that this increase correlated with an earlier age at onset. The researchers also found that longer expansions were more hypermethylated, in both a nearby CpG island and the CG-rich expanded-repeat sequences themselves. This in turn dampened expression of the C9ORF72 gene. Longer repeats therefore correlated with less C9ORF72 expression as well as with younger onset, Van Broeckhoven said. Further, Van Broekhoven showed that C9ORF72 expansions as short as 47 repeat units co-segregated with disease in two families and had the same DPR pathology as those with a longer expansion.
For another modifier of age at onset in progranulin-associated FTLD, Van Broeckhoven presented preliminary data of Eline Wauters, a Ph.D. student in her group. In the Belgian founder family who led to the identification of progranulin’s link to FTD, all carriers harbor a granulin mutation that prevents proper splicing of the gene. However, some relatives fall ill at age 45, others are unaffected at age 84. Van Broeckhoven’s group searched for loci containing variants that correlated with age at onset, and zeroed in on a 7 Mb region on chromosome 12 that accounts for a majority of the variance. Van Broeckhoven said the modifier is likely to reside within an intergenic region, where it may affect the expression of nearby or distant genes. “It could also be that this modifier only modifies age at onset in granulin mutation carriers, but maybe in C9ORF72 expansion carriers or sporadic patients as well,” she told Alzforum. “It’s best to keep an open mind.”
Newbies, Anyone?
Scientists at ICFTD were thrown a handful of new FTD-linked genes to unwrap, some of them better-vetted than others. Van Broeckhoven offered two—vacuolar protein sorting 13 homolog C (VPS13C) and filamin C (FLNC). As presented in a talk by her Ph.D. student Stéphanie Philtjens, mutations affecting the VPS13C protein emerged from whole-genome sequencing on 16 FTD patients who harbored no known pathogenic mutations. The researchers later identified 20 different missense mutations in this gene in 24 of 590 Belgian FTLD patients, absent in more than 1,300 matched controls. The researchers also showed a decreased expression of endogenous VSP13C in lymphoblast cells of mutation carriers. The normal function of the 86-exon-strong VPS13C gene is unknown. However, the closely related gene VPS13A encodes chorein, a protein known to play a role in regulating the neuronal and erythrocyte cytoskeleton, particularly actin filaments. Chorein mutations cause a rare, autosomal-recessive neurodegenerative disease, the movement disorder chorea-acanthocytosis.
The FLNC, whose protein also crosslinks actin and stabilizes the cell’s architecture, came up in a TDP-43 zebrafish model generated by Bettina Schmid and Christian Haass of the German Center of Neurodegenerative Diseases, Munich. This study reported that embryos lacking TDP-43 expressed more FLNC, and the same held true for FTLD-TDP postmortem brains (see Mar 2013 news story). Jonathan Janssens, a postdoc in Van Broeckhoven's group, sequenced FLNC in an initial group of 179 Belgian FTLD patients, and identified 11 missense mutations that did not occur in controls. Expression analysis of lymphoblast cells from mutation carriers showed that FLNC expression was reduced. Conversely, a subset of FTLD patients with GRN mutations had elevated FLNC expression in the frontal cortex. The results could suggest that altered expression of FLNC may somehow hasten FTLD via its interaction with TDP-43.
For her part, Rademakers also made a go at finding new causal genes in a family with unexplained FTD. She performed whole-exome sequencing on three affected relatives and looked for new genetic variants they had in common. She came up with a list of 27, and urged other researchers in the audience to scan their own family cohorts for these 27 in hopes of narrowing down the list. “One of these genes could very well contain the mutation that explains the disease in this family, but we have no idea which one,” Rademakers said. One promising candidate, KIF17, rose to the top of her list based on its function. KIF17 is a motor protein that shuttles a subunit of the NMDA receptor along dendrites to the synapse. The protein may therefore play a role in synaptic transmission.
John van Swieten of Erasmus Medical Center in Rotterdam, Belgium, uncovered a new gene by studying a family with a novel neurodegenerative disease. Twelve of its members dominantly inherited a syndrome marked by dementia and/or parkinsonism, and their brains were riddled with neuronal cytoplasmic inclusions of the intermediate neurofilament α-internexin. Van Swieten and colleagues performed linkage analysis and whole-exome sequencing, which produced thousands of potential variants. To whittle them down, the researchers dissected out neuronal inclusions and parsed their protein content via mass spectrometry. Combining genomics and proteomics, the researchers thus zeroed in on a missense mutation in protein kinase A type I-beta regulatory subunit (PRKAR1B), a protein the researchers found comingling with neurofilaments in the inclusions (see Wong et al., 2014). Van Swieten hypothesized that a missense mutation in the regulatory subunit could trigger mislocalization and heightened activity of the kinase, which in turn could hyper-phosphorylate neurofilaments. So far, this mutation has not turned up in people with FTD, PD, or AD, so van Swieten and colleagues have yet to determine if it is important beyond this single family (see Cohn-Hokke et al., 2014).
J. Paul Taylor of St. Jude’s Children’s Research Hospital in Memphis, Tennessee, shared striking new mechanistic findings on two mutations his lab had previously identified in people with multisystem proteinopathy (MSP) and ALS. MSPs trigger neurodegeneration as well as destruction of bone and muscle. Like people with ALS and subsets of FTLD, people with MSPs have inclusions that contain TDP-43 as well as other RNA-binding proteins that collectively belong to the family of heterogeneous nuclear ribonucleoproteins (hnRNPs). These hnRNPs contain prion-like domains that promote clustering. Taylor discovered missense mutations within the prion-like domains of two such proteins—hnRNPA1 and hnRNPA2B1—in families with MSP as well as one family with ALS (see Kim et al., 2013). Efforts to find the mutations in these particular hnRNPs in other cohorts of MSP, FTD, or ALS patients have come up empty so far (see Le Ber et al., 2014; Seelen et al., 2014). However, mutations in other hnRNPs—hnRNPDL and TIA-1—have been implicated recently in families with different forms of muscular dystrophy (see Viera et al., 2014; Hackman et al., 2013), and it is possible that others will follow.
Despite the unknown breadth of hnRNPs’ involvement across the spectrum of neurodegenerative disease, the underlying mechanisms the mutations revealed were startling. At ICFTD, Taylor presented data revealing that the hnRNPs use a so-called steric zipper motif to assemble into lipid-like droplets, which clouded up test tubes and looked like condensation under the microscope. These clusters formed rapidly in the presence of RNA, which the droplets hungrily soaked up. “This family of proteins are able to undergo spontaneous assembly into lipid-like droplets, transitioning between two distinct phases at physiological concentrations,” Taylor told the crowd. Taylor hypothesized that the clusters could act like mini-organelles, ushering RNA through key events in its life cycle within the expanse of the cytoplasm. Normally these clusters disassemble when the steric zippers unzip, but Taylor reported that the disease-associated mutations in hnRNPs prevented their disassembly and led to the formation of cytoplasmic inclusions.
How might this mechanism play out in cases of FTD/ALS where there is not an hnRNP mutation? Taylor went on to demonstrate that the same RNA granules that contained hnRNPs also contained VCP, a protein that causes FTD/ALS in mutation carriers. Depleting granules of VCP prevented their disassembly in cells, and eventually led to apoptosis and death. Taylor proposed that VCP mutations could prevent the physiological disassembly of hnRNP clusters, which would promote the formation of toxic cytoplasmic inclusions. Taylor is still determining whether these hnRNP droplets also contain TDP-43, but his findings, if confirmed, could point to fundamental cellular mechanisms that may underlie a spectrum of neurodegenerative diseases.—Jessica Shugart
van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R.
TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia.
Acta Neuropathol. 2014 Mar;127(3):397-406. Epub 2014 Jan 3
PubMed.
van Blitterswijk M, Mullen B, Wojtas A, Heckman MG, Diehl NN, Baker MC, DeJesus-Hernandez M, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Neumann M, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R.
Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene.
Mol Neurodegener. 2014 Sep 20;9:38.
PubMed.
Wong TH, Chiu WZ, Breedveld GJ, Li KW, Verkerk AJ, Hondius D, Hukema RK, Seelaar H, Frick P, Severijnen LA, Lammers GJ, Lebbink JH, van Duinen SG, Kamphorst W, Rozemuller AJ, Netherlands Brain Bank, Bakker EB, International Parkinsonism Genetics Network, Neumann M, Willemsen R, Bonifati V, Smit AB, van Swieten J.
PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology.
Brain. 2014 May;137(Pt 5):1361-73. Epub 2014 Apr 9
PubMed.
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, Maclea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP.
Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS.
Nature. 2013 Mar 28;495(7442):467-73.
PubMed.
Do MicroRNAs Cause Mayhem Across Frontotemporal Dementia Spectrum?
RNA in all its guises was a central theme at the 9th International Conference on Frontotemporal Dementias, held October 23 to 25 in Vancouver, Canada. After all, several disease-related proteins are known to make a living comingling with this nucleic acid. But beneath the waves of mainstream RNA research presented at the conference, an undercurrent of findings revealed that tinier snippets called microRNAs could have an outsize impact on disease. Researchers contend that microRNAs, which dampen the expression of their target genes, are misregulated in cells derived from FTD patients and in animal models of FTD. Changes in the levels of these tiny off switches had big consequences for the function of neurons in the models, and researchers are trying to understand exactly how the microRNAs may act in human disease across the FTD spectrum.
MicroRNAs, or miRNAs for short, were first discovered in 1993, shortly before their larger cousins, called small interfering RNAs, made their splash. But it was not until the turn of the century that the regulatory role of miRNA was fully recognized. The little ones are 21- to 23-nucleotide sequences shaped like hairpins, which block the expression of their target genes. Transcribed from within introns and intergenic regions scattered across the genome, immature “pri-miRNAs” are processed into shorter fragments by the enzymes Drosha and DGCR8 in the nucleus. The resulting “pre-miRNAs” then cross into the cytoplasm, where the enzyme Dicer sculpts them into mature miRNAs. A portion of the miRNA molecule recognizes and binds to target sequences in 3’ untranslated regions of mRNA targets, while another portion of the miRNA brings along gene-silencing machinery to do its business. Typical miRNAs can potentially latch onto hundreds of different mRNA targets; their binding can trigger the destruction of mRNAs or block their translation.
Missing Micro Manager.
Mice expressing a pathogenic mutant of CHMP2B (green, bottom panel) also express lower levels of miR-124 microRNA (red, bottom panel) than their wild-type counterparts. Proper regulation of some neurotransmitter receptors requires miR-124.
[Image courtesy of Gascon et al., Nature Medicine 2014.]
A subset of miRNAs are expressed predominantly in the brain. There, their intricately choreographed expression patterns orchestrate neural development. However, recent studies have reported that some crucial jobs of miRNAs extend into adult life, making these curious fragments potentially important for neurodegenerative disease. Specific miRNA molecules seem to be misregulated in Alzheimer’s and Parkinson’s diseases (see Qiu et al., 2014), and growing evidence implicates them in FTD/ALS as well (see Gascon and Gao, 2014).
At ICFTD, Fen-Biao Gao of the University of Massachusetts Medical School in Worcester presented new findings underscoring the role of miRNAs in FTD/ALS. A recent study from Gao’s lab had reported that expression of miR-9 was turned down in induced pluripotent stem cell (iPSC)-derived neurons from FTLD-TDP patients, who develop neuronal inclusions containing the RNA-binding protein TDP-43. MicroR-9 is expressed primarily in the brain. The consequence of miR-9’s suppression in these patients is unknown (see Zhang et al., 2013). At the meeting, Gao presented data implicating yet another miRNA in FTD: miR-124. He and his colleagues looked at miRNA expression patterns in iPSC-derived neurons from people with behavioral variant FTD (bvFTD) including C9ORF72 expansion carriers. After two months in culture, these neurons drastically downregulated expression of miR-124. This microRNA was just reported independently by another group to target the GluA2 subunit of the AMPA receptor (see Ho et al., 2014), and miR-124 binding sites reside in the 3’ UTRs of three of AMPAR’s four subunit genes. Gao’s lab found that iPSC-derived neurons from people with bvFTD expressed more of these subunits, and mutating the miR-124 binding sites in these receptor genes returned receptor levels to normal. Postmortem brain samples from people with bvFTD also had lower miR-124 and higher GluA2 expression. Overexpression of the GluA2 subunit alters the ratio of calcium-permeable and -impermeable receptor subunits, altering the AMPA receptor function. This imbalance could spell trouble for neurons, Gao said.
This AMPAR/miR-124 relationship also revealed itself in the context of an entirely different FTD mutation. A completely penetrant CHMP2B mutation identified in a single Danish family causes neuronal inclusions containing p62, but not TDP-43. Gao presented findings from a new transgenic mouse in which researchers can switch on expression of the pathogenic CHMP2B mutant gene in the brain. These mice develop deficits in social interactive behavior at a relatively young age and groom themselves obsessively when they get older. These deficits make them useful models for bvFTD, Gao said. Like their human iPSC-derived counterparts, neurons in these mice expressed less miR-124 and more of certain AMPAR subunits than controls, but delivering miR-124 into their brain using adeno-associated virus restored normal AMPAR expression levels and partially rescued the animals’ behavioral phenotype. Gao’s study came out on November 17 (see Gascon et al., 2014).
Gao believes that some FTD/ALS causal mutations may somehow converge on similar changes in miRNA expression. “MiR-124 downregulation and downstream changes in AMPAR subunits might be common to other forms of FTD,” Gao told Alzforum. “Now we will try to figure out how different disease mutations trigger changes in miRNA expression.”
Previous studies implicated TDP-43 in processing miRNAs, suggesting that the protein’s aggregation in disease would prevent the production of some miRNAs (see Feb 2012 news story). However, the way CHMP2B mutations foul up miRNA processing could involve defects in autophagy, which also has been reported to affect miRNA activity by gobbling up Dicer and other miRNA co-conspirators (see Gibbings et al., 2013).
It appears that the entire spectrum of FTLD genes may be affected by miRNA. At ICFTD, Sébastien Hébert of Laval University in Quebec presented results on tau. Spurred by findings from his and other labs that the miR132/212 family of microRNAs were downregulated in the brains of FTLD-tau patients (see Hébert et al., 2013), Hébert’s lab generated miR132/212 knockout mice. Both the human and mouse tau genes contain a miR-132/212 binding site in their 3’ UTR. The mice expressed more tau protein, which became hyperphosphorylated by six months of age and formed insoluble aggregates by 12 months. The mice had memory problems and some behavioral deficits, Hébert said, but lived a normal lifespan without obvious neurodegeneration.
Their neurons had more and larger autophagic vacuoles. Hébert speculated that the upregulation of tau could have indirectly messed up autophagy. Alternatively, other genes regulated by miR-132 could be at play, but his group has already ruled out involvement of obvious autophagy-related genes.
Hébert told Alzforum that beyond affecting brain function in wild-type mice, knocking out miR-132 also exacerbated the phenotype of FTD or AD models. For example, deleting miR-132 in a mouse expressing human tau with a pathogenic P301L mutation accelerated tauopathy. The same held true for the 3xTg mouse, which expresses mutant tau, APP, and PS1 genes. Given these findings, it is possible that miR-132 may act both as a genetic risk factor in its own right, and as a disease modifier in the presence of other, causal mutations, he said.
This is not the first time miR-132 has been in the FTLD spotlight. A previous study led by Alice Chen-Plotkin and Virginia Lee at the University of Pennsylvania in Philadelphia reported that this microRNA family shuts down expression of TMEM106B, a known genetic modifier of FTLD. Patients expressed low levels of these microRNAs, which led to boosted TMEM106B expression. TMEM106B accumulated in endolysosomes, which disrupted the trafficking of proteins including progranulin (see Aug 2012 news story). Hébert said that unlike the human TMEM106B gene, mouse TMEM106B harbors no miR-132 binding site, so he was unable to study this relationship in his knockout mice. Coming full circle, Gao and colleagues re-analyzed Chen-Plotkin’s data and found that FTD patients with granulin mutations expressed lower levels of miR-124, as well.
Still other miRNAs have been implicated in FTLD/ALS. Rosa Rademakers at the Mayo Clinic in Jacksonville, Florida, found altered expression of five different miRNAs in the cerebellum of FTLD patients with progranulin mutations, and this correlated with dysregulation of 18 genes (see Kocerha et al., 2011). Rademakers first implicated miRNAs in FTD when in 2008 she reported variation of a miRNA binding site in the progranulin gene as a risk factor (see Nov 2008 conference coverage). Other studies reported upregulation of miR-155 in patients with ALS, and treated motor defects in a mouse model with antisense oligonucleotides targeting it (see Nov 2013 conference coverage).
Hébert believes that these growing findings support the notion that the genetic influence of miRNA spreads far and wide. Like transcription factors, miRNAs are likely to play broad, important roles in many disease processes, he said. Unfortunately, most large genetic studies pay them no mind. Exome sequencing discounts them entirely, and most GWAS have not looked at SNPs that fall into miRNA sequences or promoters, or within genes that regulate miRNAs or support their function. Hébert said he hopes emerging research implicating miRNAs in disease will motivate researchers to consider miRNAs in future genetic studies.—Jessica Shugart

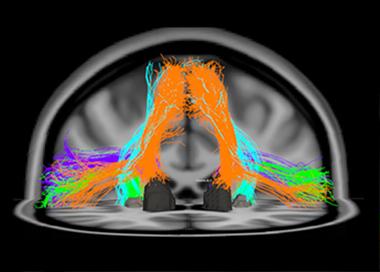
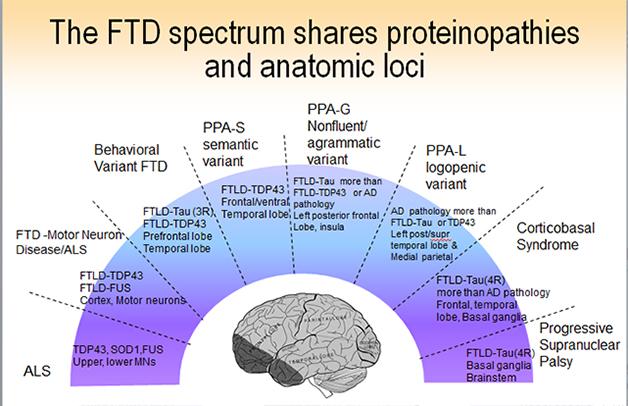
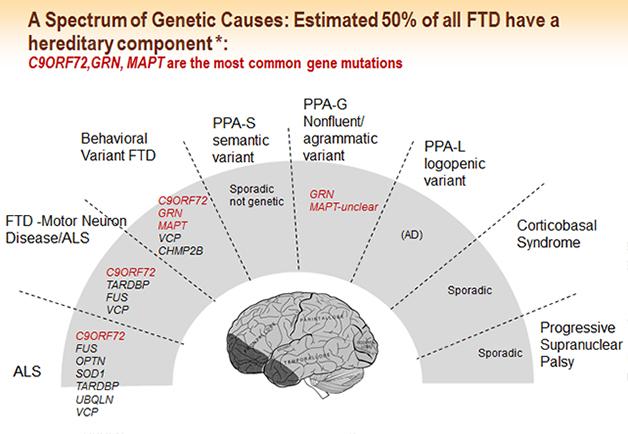
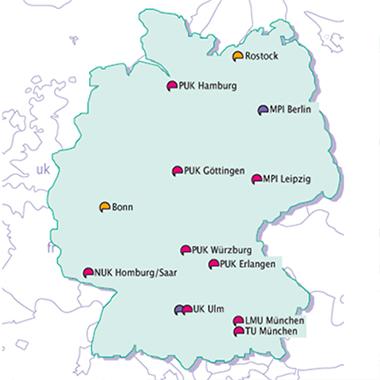



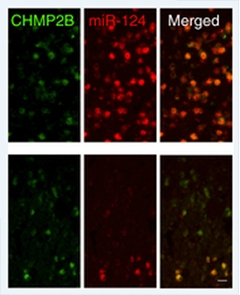
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.