Neurodegeneration—It’s Not the Tangles, It’s the T Cells
Quick Links
Here’s a tau twist that may take some adapting to. CD4+ and CD8+ T cells, yes, those mercurial executioners of the immune system, may be responsible for the neurodegeneration seen in Alzheimer’s disease and other tauopathies. That’s the scenario outlined by David Holtzman, Washington University, St. Louis, and colleagues in Nature on March 8. The scientists report that, in mice, microglia summon T cells into the brain and, perhaps by presenting antigens to them, kick them into overdrive. The cellular communication still needs to be deciphered. Even so, eliminating the T cells, or the microglia, forestalled neurodegeneration in tauopathy mice, though not in models of amyloidosis. Neuropathology data also suggest that T cells could provoke neurodegeneration in people who have AD.
- In an ApoE4/tau transgenic mouse model, T cells enter the brain.
- Some have clonally expanded in response to antigen.
- Removing these T cells averts neurodegeneration, brain atrophy.
- Does this mean Alzheimer’s is an autoimmune disease?
Holtzman hopes the findings will be a game-changer. “Of all the research that has come out of my lab, I believe this has the potential to be the most impactful,” he told Alzforum.
Other scientists were impressed. “This paper is of exceptional conceptual interest to the field of neurodegeneration because it unveils an autoimmune aspect to the neurotoxic effects of tau,” wrote Costantino Iadecola, Weill Cornell Medicine, New York (comment below). Martin Citron, UCB Pharma, Brussels, echoed Iadecola’s sentiments. “As far as I know, in the AD field over the last 30-plus years, nobody has talked about T cells in a major way. As such, the work is conceptually very exciting,” he told Alzforum. In a Nature News & Views, Ian Guldner and Tony Wyss-Coray, Stanford University, wrote that “the authors’ work implies the need for a revised view of adaptive immunity in the brain.”
T Cells in the Brain. This video shows that in a mouse model of tauopathy, T cells (green) cozy up to microglia (red) in brain tissue rather than endothelial cells (turquoise), confirming that the T cells are truly in the parenchyma, and not in the blood vessels. [Courtesy of Chen et al., Nature 2023.]
The field has been heading in this direction for some years. Long thought to be “immune privileged,” i.e., isolated from the B and T lymphocytes of the adaptive immune system, scientists have come to realize that the brain is anything but. From the meninges and bone marrow surrounding the brain, adaptive immune cells can trickle into the parenchyma (Aug 2018 news; May 2021 news; Jun 2021 news). Whether from those niches or the blood, T cells infiltrate the CSF or parenchyma of people with AD, Parkinson’s disease, and Lewy body dementia (Jan 2020 news; Amin et al., 2020; Galiano-Landeira et al., 2020). Wyss-Coray and colleagues reported that T cells cozy up to Lewy bodies, where they unleash inflammatory cytokines (Oct 2021 news). In AD, some T cells correlate with tau but not amyloid pathology, while others surround plaques (Merlini et al., 2018; Dec 2022 news).
Given this prior work, what exactly is new here? “Though we have had these papers suggesting an increase in certain T cells in the brain in AD and in primary tauopathies, nobody was thinking tauopathy was an autoimmune disease, and nobody had fully characterized what these cells were doing,” said Holtzman. Jason Ulrich and others in his lab, in collaboration with Maxim Artyomov at WashU, set out to take a comprehensive look.
First author Xiaoying Chen and colleagues began by identifying all the immune cells in the mouse brain using single-cell RNA-Seq. They used two models of amyloidosis, APP/PS1-21 and 5xFAD mice, and one of tauopathy, P301S tau, all expressing ApoE4, microglia, T cells, and B cells. The only difference Chen saw between the models was the number of T cells, which was higher in P301S/ApoE4 (TE4) mice than in APP/PS1/E4 (APE4), 5xFAD/E4 (5E4) or ApoE4 (E4) controls. Chen confirmed this by staining brain tissue for the pan T cell marker CD3 (see image below).
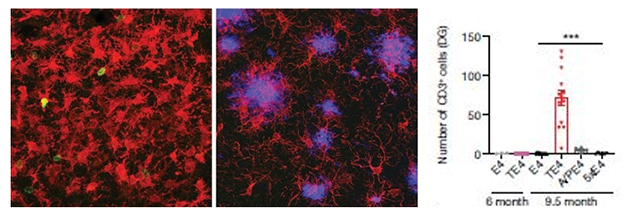
T Time. CD3+ T cells (green) infiltrate the dentate gyrus of 9.5-month-old tau/ApoE4 mice (left), but not of age-matched APP/PS1/ApoE4 mice (middle). Microglia are red, plaques purple. Quantification on right. [Courtesy of Chen et al., Nature 2023.]
Why did these T cells sneak into the brain? And what were they doing there?? Single-cell RNA-Seq was telling. Subsets of effector T cells, i.e., the ones that respond when presented with an antigen they recognize, were larger in the parenchyma, while naïve and regulatory T cells preferentially hung out in the meninges. Analysis of the T cell receptors on CD4+ and CD8+ effector cells suggested that they had expanded from single cells, an indication they had responded to an antigen.
Chen detected six types of CD4+ cells, of which two had expanded in tau/ApoE4 versus ApoE4 mice. Of 10 types of CD8+ cells, two had multiplied. Pseudotime analysis, which uses comparative transcriptomics to simulate the sequence of when different cell states emerge, suggested that unique, clonally expanded T cells were dynamically shifting from activated to “exhaustive” states. The latter indicate persistent activation by antigen, suggesting a chronic immune response in the brain. While exhaustion hampers the ability of T cells to respond, Holtzman believes these T cells can also be bad news. They behave a bit like senescent cells, sitting around provoking injury.
All told, the findings suggest that in the ApoE4 tauopathy model, T cells are being recruited to and activated in the brain, and could damage neurons. What causes this to happen is unknown, but existing hints point to microglia. For CD4+ and CD8+ T cells, activation typically begins when they interact with MHC class II and MHC class I antigen-presenting cells, respectively. This happens in lymph nodes. “Even in autoimmune diseases of the central nervous system, such as multiple sclerosis, antigen activation occurs outside the brain,” noted Holtzman.
Could tauopathy be an exception? Chen found that the increase in T cells in the parenchyma coincided with a surge in microglia expressing MHC class II proteins, and in microglia expressing CD11c, a marker of TREM2+ disease-associated microglia. Proliferation of both types has been detected in mouse models of AD and in the AD brain (Jan 2020 news; Dec 2020 news; Oct 2022 news). “Microglia are not known to be antigen-presenting cells, but they are definitely involved in homing responses, so it’s possible they may also activate the T cells,” suggested Holtzman.
In their News & Views, Guldner and Wyss-Coray didn’t rule out this idea; however, they noted that Chen and colleagues provide mostly correlative data to support it. “Perhaps there is a subset or state of microglia endowed with potent antigen-presentation skills,” Guldner and Wyss-Coray suggested. Indeed, Chen found that, in cell culture, microglia activate T cells when given interferon-γ. Immune cells produce IFNγ in a range of infectious and other sickness conditions, and scientists have detected interferon-response microglia in models of neurodegeneration and in the AD brain (Mathys 2017; Sala Frigeiro et al., 2019). Once exposed to IFNγ, the microglial activated T cells almost as well as did classic antigen-presenting cells. Amanda McQuade, University of California, San Francisco, noted that other immune signaling molecules may be involved. “The authors highlight the importance of type II interferons […], but there are many other cytokine ligand pairs that likely contribute to aging and disease including CXCL12/CXCR4 as discussed by the Wyss-Coray group and type I interferons, which I and others are investigating,” she wrote (comment below).
Does any of this implicate T cells in the killing of neurons? After all, how neurons actually perish in Alzheimer's and related diseases is a maddeningly unsolved question despite many years of research. An answer—at least for these mice—came when Chen ablated T cells by injecting tau/ApoE4 mice every five days with anti-CD4 and anti-CD8 antibodies, beginning at 6 months of age. By 9.5 months, their parenchymas were devoid of T cells. Strikingly, the pattern of phospho-tau staining placed them at a much earlier stage of disease than untreated littermates, and their microgliosis and atrophy were but a fraction (see image below). All told, the data indicate that T cells play an important role in tau-mediated neurodegeneration in these mice.

Anti-T Time. Sections from the brains of 9.5-month-old tau/ApoE4 mice show extensive atrophy (left). Fourteen weeks of anti-CD4 and anti-CD8 immunotherapy dramatically preserved their brain tissue (right). [Courtesy of Chen et al., Nature 2023.]
“This important insight could potentially lead to a paradigm shift in our understanding of how the immune system contributes to disease progression, and has the potential to lead to a differentiated set of new therapeutic strategies,” wrote Joe Lewcock, Denali Therapeutics, South San Francisco.
Previously, Michal Schwartz’s group at the Weizmann Institute of Science, Rehovot, Israel, reported that blocking immunosuppressive checkpoint proteins tempered cognitive impairment in mouse tauopathy models (Feb 2019 news). Given this new data, this might seem like it would make things worse, since it would allow the immune system to run amok; however, Chen and colleagues note that the PD-1 checkpoint Schwartz targeted also relieves suppression of regulatory T cells that keep the immune system in check. Indeed, Chen found that one week of blocking the PD-1 checkpoint in tau/ApoE4 mice drove up the number of Treg cells in the brain by 50 percent without a change in CD8+ effector cells, suggesting the checkpoint inhibitor increased only immunosuppressive cells. Chronic PD-1 blockade beginning at 8 months halved tau phosphorylation detected by the AT-8 antibody, and preserved brain volume.
Could this work in people? In samples of AD brains, Chen et al. report that the T cell count was elevated in seven people who were in Braak stage VI versus three in stages I or II, indicating immune system activation as disease progresses. More human data are needed. “We all know that mouse models and the human condition don’t always match, so we may be seeing something more dramatic in mice in gene overexpression conditions than we do in a slowly progressing disease,” Citron said. Even so, Citron believes this work opens the door to potentially treating AD with established approaches to temper certain T cells, as is done in multiple sclerosis and other conditions.
“It might not be viable to deplete entire immune-cell populations in humans, but modulating the immune response could be an alternative,” wrote Guldner and Wyss-Coray. Lewcock is of the same mind. He thinks broad immune suppression would not work and that, if microglia are the antigen-presenting cells, then the drugs would have to cross the blood-brain barrier (comment below). For Schwartz, this work signals a new era in the search for AD and tauopathy treatment, in which the targets are in the immune system (comment below).
Holtzman agrees there is still a lot to learn, including identifying the antigen-presenting cells, finding out where they present it to T cells, and figuring out what the antigen is. Is it tau itself, or some other molecule? The answers may lead to new treatment options.—Tom Fagan
References
News Citations
- Tiny Passageways Connect Skull Bone Marrow to the Brain
- As Mice Age, T Cells Traipse Around Their Meninges. Mayhem Ensues
- Private Stock—Brain Taps Skull Bone Marrow for Immune Cells
- Attack of the Clones? Memory CD8+ T Cells Stalk the AD, PD Brain
- Intruder Alert: Inflammatory T Cells Lurk Near Lewy Bodies, Neurons
- In AD, CSF Immune Cells Hint at Mounting Mayhem in the Brain
- Human and Mouse Microglia React Differently to Amyloid
- Most Detailed Look Yet at Activation States of Human Microglia
- Human Microglia Mount Multipronged Response to AD Pathology
- Cancer Treatment Takes Aim at Tauopathy
Research Models Citations
Paper Citations
- Amin J, Holmes C, Dorey RB, Tommasino E, Casal YR, Williams DM, Dupuy C, Nicoll JA, Boche D. Neuroinflammation in dementia with Lewy bodies: a human post-mortem study. Transl Psychiatry. 2020 Aug 3;10(1):267. PubMed.
- Galiano-Landeira J, Torra A, Vila M, Bové J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson's disease. Brain. 2020 Dec 1;143(12):3717-3733. PubMed.
- Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener Dis. 2018;18(1):49-56. Epub 2018 Feb 7 PubMed.
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017 Oct 10;21(2):366-380. PubMed.
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, Möller T, Hudry E, Das S, Saido T, Karran E, Hyman B, Perry VH, Fiers M, De Strooper B. The Major Risk Factors for Alzheimer's Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019 Apr 23;27(4):1293-1306.e6. PubMed.
- Gate D, Tapp E, Leventhal O, Shahid M, Nonninger TJ, Yang AC, Strempfl K, Unger MS, Fehlmann T, Oh H, Channappa D, Henderson VW, Keller A, Aigner L, Galasko DR, Davis MM, Poston KL, Wyss-Coray T. CD4+ T cells contribute to neurodegeneration in Lewy body dementia. Science. 2021 Nov 12;374(6569):868-874. Epub 2021 Oct 14 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, Wang C, Bao X, Finn MB, Hu H, Shchukina I, Kim MW, Yuede CM, Kipnis J, Artyomov MN, Ulrich JD, Holtzman DM. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023 Mar;615(7953):668-677. Epub 2023 Mar 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Weill College Medicine
This paper is of exceptional conceptual interest to the field of neurodegeneration because it unveils an autoimmune aspect to the neurotoxic effects of the tau that accumulates in the brain.
T-cells, in cross-talk with microglia, promote neurodegeneration, highlighting an interaction between innate and adaptive immunity, well known to occur in the periphery but not within the brain parenchyma.
The molecular determinants through which microglia may recruit T-cells, the antigens and related presenting cells underlying the adaptive immune response, as well as the relative contribution of microglia and T-cells to tissue damage, remain to be defined and may provide new therapeutic insights.
The role of ApoE4 in the process also remains to be elucidated. It may rely on the emerging immunoregulatory role of this apolipoprotein, which is a critical risk determinant in neurodegenerative diseases.
Overall, this is an important contribution that will inspire much new work on the immunopathology of neurodegeneration.
UCSF
This exciting work from Xiaoying Chen, David Holtzman, and colleagues highlights the growing body of work supporting T cell involvement in tauopathy and neurodegenerative disease. The authors show a specific enrichment of CD8 T cells in the parenchyma of mice with tau and APOE4 (though they note APOE4 is not necessary for this induction). It will be interesting to see if similar T cell expansions are present in other forms of tauopathy as well.
The authors highlight that depleting T cells, or targeting T cell immune checkpoints, slows neurodegeneration. Surprisingly, in amyloid models, it has been shown that genetic immunodeficiency reduces pathology and disease progression (Marsh et al., 2016). From a mechanistic standpoint, it would be interesting to see if genetic loss of T cells still ameliorates tau pathology, although the methods used by Chen et al. are more therapeutically relevant.
My personal interest lies in understanding the relationship between microglia and T cells. The authors provide good evidence that microglia are presenting antigen to T cells, as has been recognized by the Gate lab. I am looking forward to seeing future studies that investigate the specific TCRs that are expanded and uncover which antigens microglia may be presenting in various neurodegenerative diseases.
It is puzzling that loss of T cells leads to a decrease in MHCII expression on microglia—clearly, the findings in this manuscript support multiple avenues of research, including a close investigation of the relationships between peripheral immune activation and microgliosis. The authors highlight the importance of type II interferons in microglia/T cell cross talk, but there are many other cytokine ligand pairs that likely contribute to aging and disease. These include CXCL12/CXCR4 as discussed by the Wyss-Coray group (Gate et al., 2021) and type I interferons, which I and others are fervently investigating.
Looking forward, it will be important to take an unbiased approach in our studies of parenchymal immune interactions in tauopathy and to synthesize data from genetic models and pharmaceutical manipulations.
References:
Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, Fote GM, Lau L, Weinger JG, Lane TE, Inlay MA, Poon WW, Blurton-Jones M. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A. 2016 Mar 1;113(9):E1316-25. Epub 2016 Feb 16 PubMed.
Gate D, Tapp E, Leventhal O, Shahid M, Nonninger TJ, Yang AC, Strempfl K, Unger MS, Fehlmann T, Oh H, Channappa D, Henderson VW, Keller A, Aigner L, Galasko DR, Davis MM, Poston KL, Wyss-Coray T. CD4+ T cells contribute to neurodegeneration in Lewy body dementia. Science. 2021 Nov 12;374(6569):868-874. Epub 2021 Oct 14 PubMed.
Denali Therapeutics
This manuscript provides exciting new data to suggest that T-cells may contribute to neurodegeneration in Alzheimer’s disease and frontotemporal dementia. This important insight could potentially lead to a paradigm shift in our understanding of how the immune system contributes to disease progression, and has the potential to lead to a differentiated set of new therapeutic strategies.
Advances in our understanding of the human genetic contributors to Alzheimer’s disease over the past 15 years have led to an increased focus on the role of the immune system, but this focus has been largely on the role of microglia as the resident immune cells in the brain. It has been less clear whether the peripheral immune system also contributes to disease progression. Significant effort in recent years has focused on how genetic risk factors influence microglial cell state/function, and how these functions may directly contribute to degeneration.
This new paper reveals that one function of microglia may be to recruit CD8+ killer T-cells into the CNS, which subsequently drive neurodegeneration, particularly in models with tau pathology. As similar increases in T-cells were observed in brains of Alzheimer’s patients, it is possible that CNS infiltration of this cell type contributes to degeneration and functional decline in patients, as well.
As the mechanisms that underlie T-cell activation and clonal expansion in the periphery have been extensively studied, this finding has the potential to be rapidly translated into targeted therapies aimed at these mechanisms in the CNS.
Based on these results, therapeutic approaches targeting T cells are likely to capture the interest of many organizations that are working to develop treatments for Alzheimer’s and FTD, though many questions remain to be solved if this approach is to be effectively translated to the clinic. First, broad suppression of T-cell activation is not likely to be well tolerated in elderly individuals with dementia, and strategies should be investigated that are more selective for the T cell population present in the CNS.
Second, if microglia indeed act as antigen presenting cells to elicit a T cell response, then effective drugs would likely need to readily (and perhaps selectively) access the CNS in order to be effective. Lastly, biomarkers of the T-cell response that can be used for patient selection or target engagement would be required to clinically de-risk this approach prior to conducting large-scale trials. Nonetheless, this study provides an exciting start to what is sure to be an area of active investigation in the years to come.
Weizmann Institute of Science
This paper emphasizes once again a paradigm that is becoming increasing clear: different forms of dementia, including Alzheimer’s disease, are not diseases of only the brain, but also involve the immune system and the immune-brain axis. The present study further highlights the complexity of this relationship.
Specifically, the authors nicely showed that disease pathology in a new mouse model that combines tauopathy with human ApoE-4 expression is associated with detrimental microglia and cytotoxic CD8+ cells; depletion of each of these cell populations reduces disease manifestations. These findings, however, cannot be used as arguments that microglia are always and uniformly detrimental, or that T cells are uniformly harmful, and that these effects are at all stages of the disease.
The authors used anti-PD-1 antibody as a therapeutic approach, and attribute the beneficial effect of this treatment to the recruitment of regulatory T cells (Tregs) to the brain. A similar mechanism was proposed in another model of tauopathy, in which targeting the PD-1/PD-L1 pathway mitigated disease manifestations, at least in part via the recruitment of monocytes and Tregs (Ben-Yehuda et al., 2021).
The efficacy of anti-PD-1/PD-L1 immune checkpoint therapy as a treatment for dementia was also shown in models of dementia driven by amyloidosis (Baruch et al., 2016; Rosenzweig et al., 2019; Zou et al., 2021; Xing et al., 2021; Dvir-Szternfeld et al., 2022). In those studies, the effect was attributed to homing of monocyte-derived macrophages that contributed to dampening local brain inflammation. Interestingly, the same treatment was recently shown to be beneficial in mitigating natural aging-related pathology in mice, through the depletion of senescent cells in several organs (Wang et al., 2022).
Much additional work is needed to comprehensively understand the brain’s immunological mechanisms. Nevertheless, these and other recent findings demonstrate the role of systemic immune cells in controlling the development and progression of neurodegeneration.
We are entering a new era in the search for treatments for Alzheimer’s disease and tauopathy, in which the targets are not necessarily the pathological proteins themselves, but the immune system, which can regulate their accumulation in the brain, and the inflammatory damage associated with proteinopathies.
References:
Ben-Yehuda H, Arad M, Peralta Ramos JM, Sharon E, Castellani G, Ferrera S, Cahalon L, Colaiuta SP, Salame TM, Schwartz M. Key role of the CCR2-CCL2 axis in disease modification in a mouse model of tauopathy. Mol Neurodegener. 2021 Jun 25;16(1):39. PubMed.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, Kertser A, David E, Amit I, Schwartz M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. 2016 Feb;22(2):135-7. Epub 2016 Jan 18 PubMed.
Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, Keren-Shaul H, Ben-Yehuda H, Weill-Raynal P, Cahalon L, Kertser A, Baruch K, Amit I, Weiner A, Schwartz M. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun. 2019 Jan 28;10(1):465. PubMed.
Zou Y, Gan CL, Xin Z, Zhang HT, Zhang Q, Lee TH, Pan X, Chen Z. Programmed Cell Death Protein 1 Blockade Reduces Glycogen Synthase Kinase 3β Activity and Tau Hyperphosphorylation in Alzheimer's Disease Mouse Models. Front Cell Dev Biol. 2021;9:769229. Epub 2021 Dec 16 PubMed.
Xing Z, Zuo Z, Hu D, Zheng X, Wang X, Yuan L, Zhou L, Qi F, Yao Z. Influenza vaccine combined with moderate-dose PD1 blockade reduces amyloid-β accumulation and improves cognition in APP/PS1 mice. Brain Behav Immun. 2021 Jan;91:128-141. Epub 2020 Sep 19 PubMed.
Dvir-Szternfeld R, Castellani G, Arad M, Cahalon L, Phoebeluc Colaiuta S, Keren-Shaul H, Croese T, Burgaletto C, Baruch K, Ulland T, Colonna M, Weiner A, Amit I, Schwartz M. Alzheimer’s disease modification mediated by bone marrow-derived macrophages via a TREM2-independent pathway in mouse model of amyloidosis. . Nature Aging 2: Jan 2022
Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E, Imoto S, Furukawa Y, Yoshimura A, Nakanishi M. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature. 2022 Nov;611(7935):358-364. Epub 2022 Nov 2 PubMed.
TrueBinding
This interesting paper highlights that adaptive immunity plays a role in neurodegeneration, particularly Alzheimer's disease. It would be worth investigating if are there any TCRs that interact with microglial antigen-presenting cells. Is there any other antigen involved in this interaction, like TREM2? Autoinflammatory side effects are major setbacks as far as current therapies are concerned. It may be possible that removal of these T-cells either by antibody or small molecule may be therapeutic event. It would be worth investigating the interaction of T-cells with microglia in other mouse models, particularly related to Aβ.
University of Sydney
Congratulations to Xiaoying and the Holtzman lab for this very thought-provoking paper. Perhaps there could be some ideas around crossing NOD-SCID mice with tauopathy mice? The NOD-SCID mouse model has a much-weakened adaptive immune response (less T and B cells) but still has its innate immune system intact, and is typically used for cancer xenograft models. In my opinion it is a better model than the strongly immunocompromised ones such as Nu/Nu, but this could also be tried.
I would also be interested to see how tauopathy mouse models respond to biologic TNF alpha inhibitors, such as Enbrel/etanercept (soluble TNF alpha, Pfizer), Remicade/infliximab (Merck), adalimumab (Humira), golimumab (Janssen), and Cimzia/certolizumab (UCB). Some of these target insoluble TNF alpha, which may not reach certain organs and tissues, such as the iris of the eye, as readily. If epidemiological data is available, persons who have been taking these medicines could also be checked for their prevalence of AD and tauopathies.
Harvard Medical School
This is an exciting paper that suggests that microglia-T cell crosstalk may promote neurodegeneration in AD. Interestingly, this is only observed in the context of tauopathy, but not Aβ deposition.
Together with previous reports on the dysregulation of T cell responses in AD, these findings suggest that T cell-targeting therapies, alone or in combination with other approaches, may be of use for the treatment of AD. As with any other provocative findings, follow-up questions should be addressed. For example, the authors mention differences in the T cell responses detected in the parenchyma and the meninges. The authors mostly focused on the pathogenic effects of the parenchymal T cell response. However, in future studies it would be useful to interrogate the role of the meningeal response in AD pathology. In addition, a deeper mapping of the specificity of these pathogenic T cells may guide antigen-specific interventions for AD, for example based on tolerogenic nanoparticles as described for multiple sclerosis (Kenison et al., 2020).
References:
Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, Nowakowska D, Plasencia A, Stanton VP Jr, Sherr DH, Quintana FJ. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc Natl Acad Sci U S A. 2020 Dec 15;117(50):32017-32028. Epub 2020 Nov 25 PubMed.
Make a Comment
To make a comment you must login or register.