DAMned to Death? Microglia May Proliferate to Senescence
Quick Links
Overworked microglia may turn a little gray, and slow down on the job, according to new research. In the June 8 Cell Reports, researchers led by Diego Gomez-Nicola, University of Southampton, U.K., reported that in mouse models of amyloidosis and in people with Alzheimer’s, disease-associated microglia show signs of accelerated aging. These DAMs express senescence-associated genes and have shortened telomeres, even in mice as young as 10 months. Preventing microglial proliferation reduced senescent DAMs, curtailed plaques and dystrophic neurites, and preserved post-synapses. This same strategy will be tested in an upcoming clinical trial.
- Some disease-associated microglia from humans and mice replicate themselves to death.
- These senescent DAMs allow plaques to pile up and neurons suffer.
- Blocking microglial proliferation reduces plaque burden.
In the normal adult brain, microglia slowly turn over. If something goes awry, as when amyloid or tau begin to accumulate, the cells multiply in response (Aug 2017 news). But if cells rapidly divide, they run the risk of replicative senescence. With each round of mitosis, telomeres shorten, such that after about 50 cycles, they become so stubby that the cells can no longer replicate their chromosomes, they stop dividing, and enter a senescent state. This telomere-based limit was first recognized by Leonard Hayflick 60 years ago (Hayflick and Moorehead, 1961). Could microglia approach this Hayflick limit in AD?
To find out, co-first authors Yanling Hu and Gemma Fryatt turned to APP/PS1 mice, which start accumulating plaques at 4 months old. The researchers calculated how many replications it would take for microglia to achieve the cell density measured at different mouse ages. Older APP/PS1 animals hit the Hayflick limit, whereas wild-type mice did not (see image below).
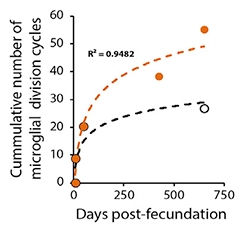
Approaching Senescence. As APP/PS1 mice age, their microglia (orange) approach the Hayflick limit of 50 cycles of cell division. Microglia in wild-type mice (black) do not. [Courtesy of Hu et al., Cell Reports, 2021.]
Were the APP/PS1 microglia senescent? In mouse hippocampal tissue, the researchers measured activity of β-galactosidase (βgal), a marker of senescence, by immunohistochemistry. Beginning when mice were 6 months old, microglial βgal activity rose along with the number of DAMs. Of the DAMs, 30 percent had high βgal activity. Using immunofluorescence, the researchers also saw that the DAMs had shortened telomeres. In fact, the length inversely correlated with expression of CD11C, a DAM signature gene.
Hu, Fryatt, and colleagues used RNA-Seq to profile the transcriptomes of DAMs and of normal microglia isolated from the APP/PS1 mice. They found 164 differently expressed genes. In addition to genes of the DAM signature, including CD11C, Clec7a, and MHCII, DAMs expressed genes previously identified in senescent cells (Hernandez-Segura et al., 2017).

DAM Senescence! Using previously published single-cell transcriptomics data from APP/PS1 mice, the researchers correlated DAM and senescence signatures. Of four microglial clusters (left), one (turquoise) expressed the DAM signature (middle). Cells in this cluster also had high senescence scores (right). [Courtesy of Hu et al., Cell Reports, 2021.]
Gene-set enrichment analysis comparing DAM gene expression to multiple published senescence gene signatures found the same. The researchers also analyzed a published single-cell microglia dataset from 16-month-old APP/PS1 mice (Van Hove et al., 2019). Again, they saw sizeable overlap between microglia expressing DAM genes and those expressing senescence markers (see image above). Taken together, the scientists concluded that DAMs show signs of senescence.
Christian Haass, DZNE, Germany, cautioned about overinterpreting these findings. "Senescent microglia may be an exhausted subpopulation of DAMs stuffed with amyloid," he told Alzforum. Marvin Reich, a student in Haass’ lab, agreed. "It is important to not equate all DAMs with senescence, but rather define senescent microglia as a population that arises from DAMs," he said.
Would preventing this senescence help or hinder the mice? To find out, the researchers fed GW2580, an inhibitor of colony-stimulating factor 1 receptor (CSF1R), to 3.5-month-old APP/PS1 and wild-type mice. CSF1R controls microglial activation and proliferation and inhibitors have been used to completely ablate the microglial population. Here, however, the authors used just enough to prevent proliferation, but not kill the existing cells.
Four months later microglial density was no higher than in wild-type mice. Few microglia were senescent, and many were not DAMs. The treated APP/PS1 mice had fewer plaques and fewer dystrophic neurites than APP/PS1 controls (see image below). Post-synapses, which degenerate in APP/PS1 mice, remained intact, as judged by levels of PSD-95.
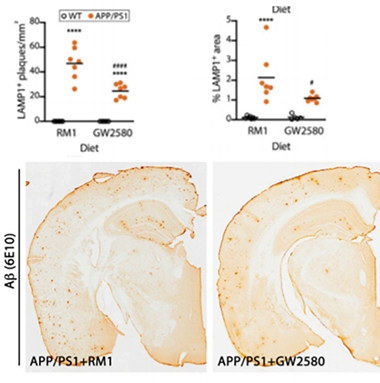
Proliferation Prevention to the Rescue. Compared to APP/PS1 mice fed a placebo diet (RM1), mice fed GW2580, which prevents microglial proliferation, had fewer senescent microglia (top, left), DAMs (top, right), and plaques (bottom). [Courtesy of Hu et al., Cell Reports, 2021.]
Lis de Weerd, also in Haass' lab, was curious about this treatment paradigm. "I expect inhibition at this level would have broad effects on microglial state, not just proliferation,” she said. “Although there might be fewer DAMs, I would like to know what kind of populations are induced and to what extent these are homeostatic."
On a similar note, Oleg Butovsky, Brigham and Women’s Hospital, Boston, suggested that GW2580 may have reduced microglial migration toward plaques rather than inhibiting their proliferation. “The authors did not measure the effect of GW2580 treatment on telomere length, which would be required to validate their hypothesis,” he wrote (full comment below).
Marco Colonna and Yingyue Zhou, Washington University, St. Louis, also wondered how plaques and senescence are linked. They noted that TREM2-negative mice have more plaques than controls, even though TREM2-negative microglia can’t proliferate or convert to DAM and so are not likely senescent. “Could the senescence phenotype seen in AD microglia be a consequence of plaque deposition and not the cause?” they wrote (full comment below).
These findings do agree with two recent studies reporting that reducing microglia numbers in 5xFAD mice limits plaque growth and preserves memory (Mar 2018 news; Sept 2019 news). In a twist on that idea, others have reported that microglia condense Aβ into dense core plaques as a way of protecting the brain from soluble toxic forms of the peptide (Apr 2021 news; Dec 2017 news).
Could preventing microglial over-proliferation be a viable AD treatment, then? Haass contrasted the protective effects of preventing microglia growth presented in this paper versus boosting their proliferation described in other studies. "Agonistic antibodies to TREM2 developed by several labs consistently stimulate proliferation and survival of microglia, and they exert clear disease ameliorating effects," he said. He thinks it may be better to target senescent cells rather than proliferating ones. "Selectively wiping out senescent microglia or rejuvenating them may be therapeutic, especially in late-stage AD when this cell population is large," he suggested.
Hu, Fryatt, and colleagues found that a group of genes called the senescence-associated secretory pattern (SASP), which includes interleukin-1β, IL-6, Caspase-8, and P16, was upregulated in the cortices of seven brain donors who had died with AD, hinting that reducing microglial senescence might work in people.
A Phase 1b trial testing Janssen’s CSF1R inhibitor Edicotinib is gearing up to recruit 54 people with mild cognitive impairment. Scientists will track changes in various CSF and blood biomarkers, as well as any adverse events. In the P301S tau mouse model of tauopathy, Edicotinib prevented microglial proliferation, which calmed spinal cord inflammation, lowered CSF phospho-tau levels, and spared motor neurons (Mancuso et al., 2019).—Chelsea Weidman Burke
References
News Citations
- Long Live the Microglia! Studies Trace Their Lifespans in Mice and Humans
- Wiping Out Microglia Prevents Neuritic Plaques
- Are Microglia Plaque Factories?
- Microglia Build Plaques to Protect the Brain
- Do Microglia Spread Aβ Plaques?
Research Models Citations
Therapeutics Citations
Paper Citations
- HAYFLICK L, MOORHEAD PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961 Dec;25:585-621. PubMed.
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol. 2017 Sep 11;27(17):2652-2660.e4. Epub 2017 Aug 30 PubMed.
- Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, Vandamme N, De Schepper S, Van Isterdael G, Scott CL, Aerts J, Berx G, Boeckxstaens GE, Vandenbroucke RE, Vereecke L, Moechars D, Guilliams M, Van Ginderachter JA, Saeys Y, Movahedi K. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019 Jun;22(6):1021-1035. Epub 2019 May 6 PubMed.
- Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzón-Sandoval J, Ribe E, Winchester L, Webber C, Nevado A, Jacobs T, Austin N, Theunis C, Grauwen K, Daniela Ruiz E, Mudher A, Vicente-Rodriguez M, Parker CA, Simmons C, Cash D, Richardson J, NIMA Consortium, Jones DN, Lovestone S, Gómez-Nicola D, Perry VH. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain. 2019 Oct 1;142(10):3243-3264. PubMed.
External Citations
Further Reading
Primary Papers
- Hu Y, Fryatt GL, Ghorbani M, Obst J, Menassa DA, Martin-Estebane M, Muntslag TA, Olmos-Alonso A, Guerrero-Carrasco M, Thomas D, Cragg MS, Gomez-Nicola D. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021 Jun 8;35(10):109228. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University School of Medicine
Washington University in St. Louis
This interesting study by Hu et al. presented two take-home messages.
First, subpopulations of AD-reactive microglia are senescent. By studying the APP/PS1 mouse model, the authors showed that activated microglia proliferate around amyloid plaques, which is consistent with our previous findings (Wang et al., 2016), such that these plaque-associated microglia reach the Hayflick limit faster than others. In line with this, the authors identified overlaps between DAM and senescence signatures, although not complete, indicating subpopulations of DAM undergo senescence. Interestingly, senescence markers were also evident in microglia from human AD samples.
Second, inhibition of microglia proliferation leads to reduced senescence in microglia, which ameliorates amyloid pathology in the APP/PS1 mouse model. The authors used a low dose of a CSF1R inhibitor that reduces microglia proliferation without killing the cells.
This paper nicely raises interest in microglia senescence in AD, a topic that has not been adequately addressed so far. One question remains to be clarified: whether reduced amyloid pathology observed in APP/PS1 treated with a low dose of CSF1R inhibitor is due to reduced microglia senescence, in other words, “rejuvenation” of microglia, or simply reduction of microglia as such, since pre-plaque depletion of microglia by a high dose of CSF1R inhibitor has been shown to prevent plaque pathology (Sosna et al., 2018; Spangenberg et al., 2019).
We noticed that Trem2-/- 5XFAD microglia have a defect in proliferation and do not convert to DAM (Wang et al., 2015), likely preventing a senescence phenotype; yet Trem2-/- 5XFAD mice develop worse pathology and microglia in these mice are more apoptotic (Ulland et al., 2017). Could the senescence phenotype seen in AD microglia be a consequence of plaque deposition and not the cause? Further studies are needed to establish a causal relationship between microglia senescence and pathology.
References:
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016 May 2;213(5):667-75. Epub 2016 Apr 18 PubMed.
Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, Glabe CG. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer's disease. Mol Neurodegener. 2018 Mar 1;13(1):11. PubMed.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, Habets G, Rymar A, Tsang G, Walters J, Nespi M, Singh P, Broome S, Ibrahim P, Zhang C, Bollag G, West BL, Green KN. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019 Aug 21;10(1):3758. PubMed.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015 Mar 12;160(6):1061-71. Epub 2015 Feb 26 PubMed.
Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell. 2017 Aug 10;170(4):649-663.e13. PubMed.
Brigham and Women's Hospital/ Harvard Medical School
One of the most important questions related to the role of DAM (MGnD) microglia is their contribution to plaque pathology and disease progression in AD. The study by Hu and colleagues is a valuable contribution to our understanding of the role of early developmental microglial changes in driving Aβ pathology and it validates the heterogeneity of DAM/MGnD microglial activation states.
The authors raised the interesting idea that a high rate of microglia proliferation associated with Aβ-plaque and DAM signature results in telomere shortening and microglia senescence. The authors demonstrated that excessive microglia replication coincides with DAM phenotype in APP/PS1 mice. A significant fraction of DAM microglia displayed a senescent profile, which was investigated by also incorporating multiple other published datasets. In human AD, microglia showed no change in morphology, but strong senescent markers as compared to non-demented controls, confirming previous studies pioneered by Wolfgang Streit and others.
To address their hypothesis, the authors used the CSF1R inhibitor GW2580 to suppress microglia proliferation. Previously, several labs had showed this specific CSF1R inhibitor did not deplete microglia but inhibited microglia proliferation. The authors concluded that the inhabitation of proliferative microglia attenuated the DAM phenotype and improved disease related to neuritic plaque pathology and protection of synapses. However, it is possible that this inhibitor may also deplete both microglia and macrophages at the same time (Chalmers et al., 2017). In addition, it is not clear whether this inhibitor may also reduce microglial migration toward plaque rather than inhibiting their proliferation. Also, the authors did not measure the effect of GW2580 treatment on telomere length, which would be required to validate their hypothesis.
Thus, future studies using specific genetic and fate-map analysis approaches, and specific modulators targeting DAM/MGnD, are required in order to determine the contribution of this important phenotype to disease pathology.
References:
Chalmers SA, Wen J, Shum J, Doerner J, Herlitz L, Putterman C. CSF-1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin Immunol. 2017 Dec;185:100-108. Epub 2016 Aug 26 PubMed.
Make a Comment
To make a comment you must login or register.