A Central Role for Prion Protein in Neurodegeneration?
Quick Links
Like Aβ oligomers, soluble aggregates of tau and α-synuclein bind to cellular prion protein, poisoning neurons, according to a study published in Acta Neuropathologica. Using a standardized procedure to produce soluble aggregates of these three proteins, researchers led by Dominic Walsh, Brigham and Women’s Hospital, Boston, report that all three types of aggregate bind to the same sequence in cellular prion protein (PrPC). As a result, neurites shrank, and synapses became less plastic. “This work clearly suggests that inhibition of oligomer-to-PrPC binding is an important therapeutic target for multiple neurodegenerative diseases,” wrote Thomas Wisniewski, New York University School of Medicine.
- Soluble aggregates, not monomers, of Aβ, tau, and α-synuclein bind to PrP.
- Binding causes neurotoxicity.
- PrP may be a therapeutic target for several late-life neurodegenerative diseases.
To take things one step further, the researchers recapitulated the experiments with human material, isolating Aβ, tau, and α-synuclein oligomers postmortem from people who had had Alzheimer’s or Pick’s disease, or dementia with Lewy bodies. Extracts from all three diseases were toxic to induced human neurons, but not if the neurons were missing PrP. “Our findings suggest that both the synthetic and brain-derived aggregates of these proteins cause toxicity that requires the expression of PrP,” Walsh told Alzforum.
The study was published last December around the same time as two others implicating PrP in AD. Researchers led by Tara Spires-Jones at the University of Edinburgh reported that mice overexpressing amyloid precursor protein (APP) and tau upregulated PrP (Pickett et al., 2019).
Dietmar Thal and colleagues at KU Leuven, Belgium, demonstrated that soluble Aβ and phosphorylated tau (p-tau) interact with PrPC in vitro, in transgenic mice, and in Alzheimer’s disease brain (Gomes et al., 2019). And in mice that overexpressed soluble Aβ, its binding to PrP correlated with Aβ-driven acceleration of tangle pathology through the brain. “The more Aβ we have, the more likely we have interactions that exacerbate tau pathology,” Thal told Alzforum.
“The new findings by Walsh and colleagues support the role of PrP as a ‘general receptor’ of soluble protein aggregates,” wrote Thal and Luis Gomes, also from KU Leuven, in a comment to Alzforum (see below). “The binding of these aggregates also caused functional and structural deficits that were rescued when PrP was either ablated or blocked,” they noted.
Walsh’s study builds on previous work describing PrP as a critical Aβ oligomer receptor (Salazar and Strittmatter, 2017). “By and large, nearly every previous study has found that certain Aβ assemblies bind to PrP. Everyone agrees. Where people are disagreeing is on the consequences,” said Walsh. What’s more, not all forms of Aβ bind to PrP, he added, and some forms that do not bind might still be toxic. To complicate matters, while other studies indicate that PrP also binds tau and α-synuclein, no one has directly compared the interaction of all three with PrP (Ferreira et al., 2017; Hu et al., 2018; Ondrejcak et al., 2018). This is partly because, until now, there was no standardized procedure for preparing soluble protein aggregates, Walsh explained.
First author Grant Corbett and colleagues developed a reproducible procedure to generate homogenous soluble protein aggregates of Aβ, tau, and α-synuclein. First, they created insoluble, end-stage fibrillar aggregates from monomers of each, following established protocols (Buell et al., 2014; Hellstrand et al., 2010; O’Dowd et al., 2012). Next, they centrifuged the fibrils, removed the monomers, resuspended the fibrils, and then broke them into small soluble aggregates by sonication. Then they tested how each bound PrP and curtailed neurite growth of mouse primary neurons (MPN), and of human neurons derived from induced pluripotent stem-cells (iNs).
Aβ, tau, and α-synuclein monomers were not toxic to these cells, but soluble aggregates of all three proteins stunted neurites in both. This toxicity was PrP-dependent: Mouse and human neurons that lacked the prion protein showed no signs of neurite damage.
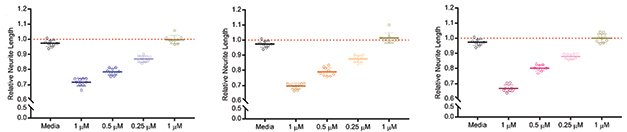
Neurotoxicity. Soluble aggregates of Aβ, tau, and α-synuclein poison mouse primary neurons in a dose-dependent manner; monomers do not. [Courtesy of Corbett et al., Acta Neuropathologica, 2019.]
Would soluble aggregates found in diseased human brains do the same? To find out, the researchers incubated induced neurons with aqueous extracts containing Aβ, tau, or α-synuclein from two brains each of people who had died with Alzheimer’s, Pick’s, and DLB, respectively. For comparison, they used AD, Pick’s, and DLB extracts from which they had depleted these aggregates, as well as extracts from one healthy brain.
All extracts from diseased brains reduced neurite length, whereas depleted extracts did not. Strikingly, neurons that had their PrP gene knocked out by CRISPR were protected from Aβ, tau, and α-synuclein aggregates, pointing once again toward a role for PrP in toxicity.

PrP Dependence. Soluble extracts (Mock) from AD, DLB, and Pick’s brains shortened neurites of induced human neurons. Neurons lacking PrP were protected (gray shading). Extracts immunodepleted using antibodies to Aβ (AW7 ID), α-synuclein (2F112 ID), and tau (Tau5 ID) were neutralized. [Courtesy of Corbett et al., Acta Neuropathologica, 2019.]
The researchers also tested how recombinant soluble protein aggregates would affect synaptic plasticity using multielectrode recordings from hippocampal slices of both wild-type and genetically ablated PrP-deficient mice. Aβ, tau, and α-synuclein aggregates all inhibited long-term potentiation (LTP), but only when the mice expressed PrP. “It looks like the PrP receptor is the target of the protein aggregates. Then, regardless of which protein, they may lead to neurodegeneration,” Thal said. Curiously, tau aggregates blocked LTP at much lower concentrations than did Aβ or α-synuclein aggregates.
Susan Catalano, Cognition Therapeutics Inc., Pittsburgh, called this difference striking, noting that the synthetic oligomers contain β-sheet conformations. “A characteristic of β-sheet structures is promiscuous unsaturable binding, usually to negatively charged residues on a variety of extracellular matrix molecules on the cell surface, including prion and gangliosides. In this light, the dramatic difference in concentrations of tau protofibrils required to block LTP compared to Aβ and α-synuclein aggregates is noteworthy,” she wrote (Matsuzaki et al., 2018).
The paper raises questions about how Aβ and tau contribute to AD pathology. “The authors immunodepleted Aβ [from AD brain extracts] with the AW7 antibody, which seemed sufficient to rescue the toxic effects. However, those brain extracts still contained tau that apparently did not cause toxicity,” Thal and Gomes wrote.
The lack of tau toxicity in these extracts puzzled Walsh, as well. “In AD, there are two hallmark proteins, Aβ and tau, but the toxicity we saw in experiments with AD brain extracts was mediated only by Aβ, not tau,” he said. “It is not clear to me what the difference is between the tau in the AD brain extract versus the tau in the Pick’s brain extract. I can only speculate that different structures may be involved.”
Researchers have found slight differences in the structures of tau fibrils isolated from people who had AD, Pick’s, and even chronic traumatic encephalopathy (Fitzpatrick et al., 2017; Falcon et al., 2018; Falcon et al., 2019).
Tiago Outeiro, University Medical Center Göttingen in Germany, thinks that from a therapeutic standpoint targeting PrP could offer a one-stop solution. “This study supports the idea that targeting PrP may constitute a valid strategy for protecting neuronal function from a series of proteotoxic injuries,” Outeiro wrote. Walsh agreed, but cautioned that the soluble aggregates are pleomorphic, and that PrP is probably not responsible for all their toxic interactions.
And how does PrP mediate toxicity? Does it promote templated misfolding of Aβ, tau, and α-synuclein, as it does for prions? Or does it act like a cell surface receptor, transducing signals to the cytosol? “We can say at the moment that PrP is involved in the process, but whether it triggers aggregation is unclear,” Thal told Alzforum. Walsh doubts PrP seeds misfolding. “It would have to bind monomer, and it does so only when monomer concentration is high enough for spontaneous aggregation,” he said.
Researchers agree on what ought to come next. “The next big step will be to examine these interactions in animal models in more detail,” wrote Colin Masters, University of Melbourne. He wants to see how these proteins interact at a molecular level. “There is still a big gap in demonstrating a biologically relevant, direct physical interaction between these extracellular and intracellular molecular entities,” he wrote. “If PrP mediates the Aβ-tau phenomenon in Alzheimer’s disease, the most parsimonious way to demonstrate this might lie in a therapeutic strategy aimed at targeting PrP in AD, and that is doable,” Masters believes.—Sandra Blumenrath
References
Paper Citations
- Pickett EK, Herrmann AG, McQueen J, Abt K, Dando O, Tulloch J, Jain P, Dunnett S, Sohrabi S, Fjeldstad MP, Calkin W, Murison L, Jackson RJ, Tzioras M, Stevenson A, d'Orange M, Hooley M, Davies C, Colom-Cadena M, Anton-Fernandez A, King D, Oren I, Rose J, McKenzie CA, Allison E, Smith C, Hardt O, Henstridge CM, Hardingham GE, Spires-Jones TL. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer's Disease. Cell Rep. 2019 Dec 10;29(11):3592-3604.e5. PubMed.
- Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CA, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019 Dec;138(6):913-941. Epub 2019 Aug 14 PubMed.
- Salazar SV, Strittmatter SM. Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer's disease. Biochem Biophys Res Commun. 2017 Feb 19;483(4):1143-1147. Epub 2016 Sep 14 PubMed.
- Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF. α-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017 Nov;20(11):1569-1579. Epub 2017 Sep 25 PubMed.
- Hu NW, Corbett GT, Moore S, Klyubin I, O'Malley TT, Walsh DM, Livesey FJ, Rowan MJ. Extracellular Forms of Aβ and Tau from iPSC Models of Alzheimer's Disease Disrupt Synaptic Plasticity. Cell Rep. 2018 May 15;23(7):1932-1938. PubMed.
- Ondrejcak T, Klyubin I, Corbett GT, Fraser G, Hong W, Mably AJ, Gardener M, Hammersley J, Perkinton MS, Billinton A, Walsh DM, Rowan MJ. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J Neurosci. 2018 Dec 12;38(50):10595-10606. Epub 2018 Oct 24 PubMed.
- Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TP, Linse S, Dobson CM. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci U S A. 2014 May 27;111(21):7671-6. Epub 2014 May 9 PubMed.
- Hellstrand E, Boland B, Walsh DM, Linse S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem Neurosci. 2010 Jan 20;1(1):13-8. PubMed.
- O'Dowd ST, Ardah MT, Johansson P, Lomakin A, Benedek GB, Roberts KA, Cummins G, El Agnaf OM, Svensson J, Zetterberg H, Lynch T, Walsh DM. The ELISA-Measured Increase in Cerebrospinal Fluid Tau that Discriminates Alzheimer's Disease from other Neurodegenerative Disorders is not Attributable to Differential Recognition of Tau Assembly Forms. J Alzheimers Dis. 2012 Oct 3; PubMed.
- Matsuzaki K, Kato K, Yanagisawa K. Ganglioside-Mediated Assembly of Amyloid β-Protein: Roles in Alzheimer's Disease. Prog Mol Biol Transl Sci. 2018;156:413-434. Epub 2018 Feb 1 PubMed.
- Fitzpatrick AW, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SH. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. 2017 Jul 13;547(7662):185-190. Epub 2017 Jul 5 PubMed.
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SH, Goedert M. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. 2018 Sep;561(7721):137-140. Epub 2018 Aug 29 PubMed.
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, Scheres SH. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019 Apr;568(7752):420-423. Epub 2019 Mar 20 PubMed.
Further Reading
News
- Right Turn: Aβ Fibril Structure from Alzheimer’s Brain Reveals Surprising Twist
- Does Tau Kill Neurons by Way of Necroptosis?
- Forget Fibrils: Lewy Pathology Is More Lipid Than Protein
- Tau Natively Unstructured? Not Always, New Study Says
- Shape of α-Synuclein Aggregates Influences Pathology
- Electron Microscope Yields Finer Structure of α-Synuclein, Aβ Fibrils
- Research Brief: Assessing Aβ Oligomers Toxicity in Live Mice
Primary Papers
- Corbett GT, Wang Z, Hong W, Colom-Cadena M, Rose J, Liao M, Asfaw A, Hall TC, Ding L, DeSousa A, Frosch MP, Collinge J, Harris DA, Perkinton MS, Spires-Jones TL, Young-Pearse TL, Billinton A, Walsh DM. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020 Mar;139(3):503-526. Epub 2019 Dec 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Katholieke Universiteit Leuven, Department of Imaging and Pathology, Laboratory of Neuropathology
KU Leuven
Since the Strittmatter group described the interaction between cellular prion protein (PrPC) and Aβ oligomers (Laurén et al., 2009), PrPC has been suggested as a mediator of functional and toxic deficits in Alzheimer’s disease. Later papers have extended this knowledge by demonstrating that PrPC is also involved in the synaptic dysfunction caused by pathological α-synuclein and tau species (Ferreira et al., 2017; Ondrejcak et al., 2018).
Within the last six months, three articles published almost simultaneously definitely placed PrPC as a central player in a number of neurodegenerative diseases. First, we demonstrated that Aβ and p-tau interact with PrPC in vitro, in transgenic mice, and in the human AD brain (Gomes et al., 2019). Moreover, we showed increased Aβ-PrPC and p-tau-PrPC interaction in APP23xTAU58 mouse models compared to TAU58 transgenic mice, which was associated with an acceleration of tau spreading, presumably induced by Aβ. Consistently, Pickett et al. found PrPC as the largest upregulated synaptic mRNA in APP/PS1+Tau mice (Pickett et al., 2019).
In this recent article, Corbett et al. support the concept of PrPC as a general receptor of soluble protein aggregates (SPAs) by demonstrating binding of PrPC to Aβ, α-syn, and tau in vitro and in vivo. Furthermore, the authors described that SPAs caused functional and toxic deficits that were rescued when PrPC was ablated or blocked by PrPC antibodies targeting the SPA’s binding sites identified in this study. In an all-human experimental paradigm using human iPSC-derived neurons, human brain extracts from AD, DLB, and Pick’s disease cases were toxic to neurons in a PrPC-dependent manner. Immunodepletion of Aβ (AD), α-syn (DLB), and tau (Pick’s disease) from the brain extracts prevented the toxic effects.
Nevertheless, the question about the relative contribution of Aβ and/or tau for the toxicity mediated by PrPC in AD cases remains open. In the two AD cases used by Corbett et al., the authors immunodepleted Aβ with the AW7 antibody, which seemed to be sufficient to rescue the toxic effects. However, those brain extracts still contained tau that apparently did not cause toxicity. On the other hand, a previous article from the same group reported one AD case in which the Tau5 antibody rescued synaptotoxicity in an Aβ-independent manner (Ondrejcak et al., 2018).
Another interesting point is the composition of the active tau species binding to PrPC. In this paper, the authors successfully use an anti-total tau antibody (Tau5) for immunodepletion of tau in Pick’s disease cases. Together with our observation that p-tau (PHF1) derived from human AD brain binds to PrPC, it is tempting to speculate that the relevant tau species might be phosphorylated and include the mid-region and c-terminal part of tau (aa 210-404).
Taken together, these findings place PrPC as a mediator of toxicity induced by pathological protein aggregates. Therefore, a possible therapy targeting PrPC could be effective for a broad range of neurodegenerative diseases that involve the formation of protein aggregates. Nevertheless, further studies using transgenic mice to explore the value of targeting PrPC genetically or pharmacologically are essential.
References:
Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009 Feb 26;457(7233):1128-32. PubMed.
Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF. α-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017 Nov;20(11):1569-1579. Epub 2017 Sep 25 PubMed.
Ondrejcak T, Klyubin I, Corbett GT, Fraser G, Hong W, Mably AJ, Gardener M, Hammersley J, Perkinton MS, Billinton A, Walsh DM, Rowan MJ. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J Neurosci. 2018 Dec 12;38(50):10595-10606. Epub 2018 Oct 24 PubMed.
Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CA, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019 Dec;138(6):913-941. Epub 2019 Aug 14 PubMed.
Pickett EK, Herrmann AG, McQueen J, Abt K, Dando O, Tulloch J, Jain P, Dunnett S, Sohrabi S, Fjeldstad MP, Calkin W, Murison L, Jackson RJ, Tzioras M, Stevenson A, d'Orange M, Hooley M, Davies C, Colom-Cadena M, Anton-Fernandez A, King D, Oren I, Rose J, McKenzie CA, Allison E, Smith C, Hardt O, Henstridge CM, Hardingham GE, Spires-Jones TL. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer's Disease. Cell Rep. 2019 Dec 10;29(11):3592-3604.e5. PubMed.
University Medical Center Goettingen
Neuronal dysfunction is thought to precede cell death in most, if not all, neurodegenerative diseases. However, it is still unclear what are the molecular events triggering neuronal dysfunction. While protein aggregation is a pathological hallmark in AD, PD, and other disorders, accumulating evidence suggests that smaller, perhaps more soluble, protein assemblies might actually be more toxic to neurons than larger protein aggregates. In this context, oligomeric species of α-synuclein (αSyn), tau, or amyloid-β have consistently been found to be more toxic that insoluble protein assemblies. Previously, different studies already suggested that the prion protein (PrP) could work as a “sensor” of certain oligomeric forms of amyloid-β peptide, αSyn, and tau, and that deletion of PrP, or pharmacological blockade using PrP antibodies, could prevent neuronal dysfunction. In particular, our group showed this for αSyn, and revealed a signaling cascade triggered by the interaction between recombinant αSyn oligomers and PrP (Ferreira et al., 2017).
In this recent study, Corbett and colleagues confirmed and expanded on previous findings, showing that soluble protein aggregates purified from human brain tissue interact with PrP, and induce synaptic dysfunction in a PrP-dependent manner. Whether PrP signals the toxicity of the three different proteins tested in the same way was not investigated. It is also still unclear whether PrP affects the conformational conversion of otherwise innocuous proteins into toxic confirmations, so this will have to be further investigated. However, this study supports the idea that some proteins, such as PrP, may sense specific protein assemblies and transduce their toxicity and that, perhaps, targeting PrP may constitute a valid strategy from protecting neuronal function from a series of proteotoxic injuries.
In the future, we will need to further detail the molecular interplay between PrP and the various proteins, assessing possible mutual effects at the structural level. In vivo studies in animal models will also be important to further document the effects described, since there is still controversy in the field.
Overall, this is an exciting study in the sense that it tells us that there may be more therapeutic options that those that meet the eye.
References:
Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF. α-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017 Nov;20(11):1569-1579. Epub 2017 Sep 25 PubMed.
Cognition Therapeutics Inc.
This comprehensive work extends previous work describing the critical role of cellular prion protein (PrP) as a member of the multiprotein Aβ oligomer receptor complex pioneered by Strittmatter and colleagues (Smith et al., 2019). As in previous published studies, knockout of prion protein reduces oligomer binding to neurons by 50 percent, and reduces toxicity. Elegant array tomography studies demonstrate that Aβ, α-synuclein, and tau co-localize with PrP at synapses in end-stage diseased human postmortem brain and potentially interact there.
The rigorous protocol used here to make uniform preparations of Aβ, α-synuclein, and tau protofibrils generates species that, while soluble, contain substantial β-sheet structure. β-Sheet conformations of a large number of proteins (including prion itself) can act as a template, triggering intrinsically disordered proteins (such as prion itself) to form more β-sheet in a process known as template-mediated conformational propagation. A characteristic of β-sheet structures is promiscuous unsaturable binding, usually to negatively charged residues on a variety of extracellular matrix molecules on the cell surface (also including prion and gangliosides; Matsuzaki et al., 2018). In this light, the dramatic difference in concentrations of tau protofibrils required to block TBS-induced LTP compared to Aβ and α-synuclein aggregates is noteworthy.
This work sets the stage for investigations of how molecular interactions of tau and α-synuclein protofibrils with PrP relate to the well-characterized interactions of Aβ and PrP with metabotropic glutamate receptors and related downstream signaling described by a number of labs, as well as the common mechanisms underlying inflammatory responses in both Alzheimer’s and prion diseases (Combs et al., 1999).
References:
Matsuzaki K, Kato K, Yanagisawa K. Ganglioside-Mediated Assembly of Amyloid β-Protein: Roles in Alzheimer's Disease. Prog Mol Biol Transl Sci. 2018;156:413-434. Epub 2018 Feb 1 PubMed.
Smith LM, Kostylev MA, Lee S, Strittmatter SM. Systematic and standardized comparison of reported amyloid-β receptors for sufficiency, affinity, and Alzheimer's disease relevance. J Biol Chem. 2019 Apr 12;294(15):6042-6053. Epub 2019 Feb 20 PubMed.
Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999 Feb 1;19(3):928-39. PubMed.
University of Melbourne
Apart from the topological problem (i.e., the cellular compartments in which each of the players exist: e.g., does extracellular PrP ever normally come into direct contact with cytoplasmic α-syn?), the biological relevance of these interactions remains poorly understood. This current paper goes a long way to increase our knowledge based on in vitro and cell-based assays. The next big step will be to examine these interactions in animal models in more detail.
As pointed out by the authors, we know from human disease studies that Aβ and PrP can drive both tau and α-syn aggregation: but there is still a big gap in demonstrating a biologically relevant direct physical interaction between these extracellular and intracellular molecular entities. If PrP mediates the Aβ-tau phenomenon in Alzheimer’s disease, the most parsimonious way to demonstrate this might lie in a therapeutic strategy aimed at targeting PrP in AD. It’s do-able.
New York University
In this elegant and important study, Walsh and colleagues demonstrate, using a variety of complementary methodologies, that PrPC is critical for mediating neurotoxicity related to soluble oligomeric species of amyloid-β, α-syn, and p-tau. They show that soluble oligomer species of each of these proteins (and not monomers or fibrils) bind to PrP at two sites and that this interaction is needed for mediating disruption of LTP and neuritotoxicity. Complementary data of specific binding is provided using primary mouse neurons, iPSC-derived human neurons, and array tomography of diseased brains. This work clearly suggests that inhibition of the oligomer to PrPC binding is an important therapeutic target for multiple neurodegenerative diseases.
This hypothesis is fully consistent with prior work from a number of groups including some of our own prior studies. We and others have shown that antibodies that block the binding of PrPSc to PrPC (at binding Site II discussed in the Walsh paper) is therapeutically efficacious in prion infection using tissue culture and in vivo models (Sadowski et al., 2009). We have also shown that in an AD model, even with advanced amyloid plaque pathology, blocking this interaction is able to produce significant cognitive benefits, even when the amyloid burden and Aβ oligomer concentration is not reduced, highlighting the potential power of blocking Aβ oligomer toxicity via its PrPC binding (Chung et al., 2010). Extending the disease-targeting spectrum to chronic traumatic encephalopathy (CTE), we have also shown that expression of PrPC is essential for the development of p-tau pathology following closed head traumatic brain injury (Rubenstein et al., 2017). Hence the targeting of PrPC could be effective in AD, prion diseases, FTD and CTE. (Wisniewski T, Boutajangout A, inventors; New York University assignee. Monoclonal antibody to treat Alzheimer's disease, prion disease, frontotemporal dementias and traumatic brain injury/chronic traumatic encephalopathy. USA2019. Application Number: 16031418.)
References:
Sadowski MJ, Pankiewicz J, Prelli F, Scholtzova H, Spinner DS, Kascsak RB, Kascsak RJ, Wisniewski T. Anti-PrP Mab 6D11 suppresses PrP(Sc) replication in prion infected myeloid precursor line FDC-P1/22L and in the lymphoreticular system in vivo. Neurobiol Dis. 2009 May;34(2):267-78. PubMed.
Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. PubMed.
Rubenstein R, Chang B, Grinkina N, Drummond E, Davies P, Ruditzky M, Sharma D, Wang K, Wisniewski T. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathol Commun. 2017 Apr 18;5(1):30. PubMed.
Istituto di Ricerche Framacologiche "Mario Negri"
This paper by Corbett et al. is an interesting piece of science related to the possible key role played by cellular prion protein (PrPc) in the neurotoxicity exerted by oligomeric forms of amyloid-β, α-synuclein, and tau, which have pathogenic roles in Alzheimer’s, Parkinson’s, and tauopathies. The role of soluble aggregates, aka oligomers, as primary culprits in protein misfolding in neurodegenerative disorders is now well-established. We coined the term “oligomeropathies” to underline the importance of this form in inducing neuronal dysfunction (Forloni et al., 2016).
The functional relevance of binding of Aβ oligomers to PrPc was initially proposed by Strittmatter’s group 10 years ago (Laurén et al., 2009). We quickly confirmed high-affinity binding between PrPc-and Aβ but found no direct functional consequences (Balducci et al., 2010). We demonstrated that PrP-knockout mice were equally as sensitive to the detrimental effect of Aβ oligomers as were wild-type animals. In the last decade, several papers have been published supporting one or the other point of view and Purro and colleagues have nicely summarized the debate (Purro et al., 2018).
The interaction of α-syn oligomers with PrPc was more recently proposed (Ferreira et al., 2017). In contrast with previous results with Aβ oligomers, in this case we and others were not able to replicate even the simple binding between PrPc and α-syn oligomers (La Vitola et al., 2019; Shirasaka et al., 2020).
This long introduction was necessary to contextualize Corbett et al.’s results, which strongly support a central role for PrPc in mediating the neurotoxic effects of oligomers originating from Aβ, α-syn, and tau fibrils. One point to consider is that these preparations, unlike others, were produced by sonication of preformed fibrils. These preparations bound PrPc, primary murine neurons, and mediated neurotoxicity, and inhibited long-term potentiation (LTP). Generally, other preparations have been made by interrupting the self-aggregation process by diluting to prevent further aggregation. This methodological difference, as pointed out by the authors, could explain the binding capacity of α-syn oligomers and the contradictory results.
The authors found that protofibrils were the conformation state active on PrPc but could not show that other oligomer conformations were active. Further studies in this vein would be appreciated. When we tested PrPc-Aβ oligomer interactions, we tried to replicate exactly the oligomer preparations described by previous studies to ensure consistency.
More impressive results are reported in the second part of the study, which showed that water-soluble extracts from AD, DLB, and PiD brains induced neurotoxic effects dependent on the presence of specific protein aggregates and in all cases the toxicity required PrP (fig.8). Immunoprecipitation of single protein Aβ from AD brain extracts, α-syn for DLB, and tau from PiD nullified the respective toxicity and the elimination of PrP from the neuronal cells (iPS-derived) completely antagonized the toxicity, as well. Furthermore, extracts of brain free from neurodegeneration was not toxic at all. In this context, it is surprising to selectively eliminate toxicity by immunoprecipitating a single molecule, when we know that aggregates of Aβ, α-syn, and tau have been found in various amounts in all three neurodegenerative disorders.
This complex study was well-designed and the interaction of various oligomers with PrPc was investigated in several ways. Unfortunately, the complexity of the assays does not favor easy replication of the experiment’s results, although their robustness overshadows methodological impediments. As indicated above, the data on PrPc-Aβ oligomer interactions are numerous. We knew little about the interaction of PrPc with α-syn oligomers or tau oligomers until now. Ferreira et al. proposed that PrPc-α-syn oligomers activated a similar intracellular pathway as did Aβ oligomers, involving metabotropic glutamate receptor 5 receptors and fyn kinase (Um et al., 2013). A clinical trial tested a fyn kinase inhibitor supposedly activated by Aβ oligomers through PrPc; unfortunately this treatment did not affect AD progression (van Dyck et al., 2019). However, as reported by the authors in the present paper and in agreement with others (Purro et al., 2018; Forloni et al., 2013; Smith and Strittmatter, 2017; Brás et al., 2018), it is reasonable to conclude that only a part of oligomer toxicity is governed by PrPc. Independently of the role of defined receptor, or simply acceptor, exerted by PrPc, its capacity to bind the oligomeric species suggests possible therapeutic strategies that need to be tested under appropriate conditions. The molecular basis of the PrPc-oligomeric interaction and the biological mechanism responsible for the toxic effect, as well as the possible competition of the different oligomers for PrPc domains, have not been investigated in this paper. Thus, several other studies are necessary to confirm the results and to find a molecular explanation for them and, importantly, to understand the biological relevance in the pathological conditions. The structure of PrPc might explain its affinity for oligomeric species, although whether a single protein can be crucial for four different neurodegenerative disorders, including prion-related encephalopathies, remains an open question.
(I have the responsibility of the text, but the content is also the fruit of discussion with my colleagues: Drs. Balducci C, Chiesa, R, Gobbi, M, La Vitola P, and Zanier E.)
References:
Forloni G, Artuso V, La Vitola P, Balducci C. Oligomeropathies and pathogenesis of Alzheimer and Parkinson's diseases. Mov Disord. 2016 Jun;31(6):771-81. Epub 2016 Mar 31 PubMed.
Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009 Feb 26;457(7233):1128-32. PubMed.
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010 Feb 2;107(5):2295-300. PubMed.
Purro SA, Nicoll AJ, Collinge J. Prion Protein as a Toxic Acceptor of Amyloid-β Oligomers. Biol Psychiatry. 2018 Feb 15;83(4):358-368. Epub 2017 Nov 21 PubMed.
Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF. α-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017 Nov;20(11):1569-1579. Epub 2017 Sep 25 PubMed.
La Vitola P, Beeg M, Balducci C, Santamaria G, Restelli E, Colombo L, Caldinelli L, Pollegioni L, Gobbi M, Chiesa R, Forloni G. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain. 2019 Feb 1;142(2):249-254. PubMed.
Shirasaka M, Kuwata K, Honda R. α-Synuclein chaperone suppresses nucleation and amyloidogenesis of prion protein. Biochem Biophys Res Commun. 2020 Jan 1;521(1):259-264. Epub 2019 Oct 18 PubMed.
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron. 2013 Sep 4;79(5):887-902. PubMed.
van Dyck CH, Nygaard HB, Chen K, Donohue MC, Raman R, Rissman RA, Brewer JB, Koeppe RA, Chow TW, Rafii MS, Gessert D, Choi J, Turner RS, Kaye JA, Gale SA, Reiman EM, Aisen PS, Strittmatter SM. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2019 Jul 22; PubMed.
Forloni G, Sclip A, Borsello T, Balducci C. The neurodegeneration in Alzheimer disease and the prion protein. Prion. 2013 Jan-Feb;7(1):60-5. PubMed.
Smith LM, Strittmatter SM. Binding Sites for Amyloid-β Oligomers and Synaptic Toxicity. Cold Spring Harb Perspect Med. 2017 May 1;7(5) PubMed.
Brás IC, Lopes LV, Outeiro TF. Sensing α-Synuclein From the Outside via the Prion Protein: Implications for Neurodegeneration. Mov Disord. 2018 Nov;33(11):1675-1684. Epub 2018 Nov 13 PubMed.
The University of Edinburgh
Further evidence of the critical role of the cellular prion protein in neurodegenerative disorders. This evidence, showing PrPc acting as a receptor for other misfolded proteins, reveals a direct link between these misfolded proteins and synaptic degeneration, due to the critical role of PrPc in sorting proteins for synaptic vesicle recycling (Peggion et al., 2018) versus lysosomal degradation (Heisler et al., 2018).
Blocking this interaction by removing PrPc as a potential therapy should be approached with caution, as it would reduce vesicle recycling and synaptic transmission. This interaction may prove to be a useful tool for targeting other misfolded proteins to lysosomal degradation. Perhaps at earlier stages of disease this may prevent extracellular spread of these misfolded proteins?
During prion disease, vesicular trafficking of proteins is disturbed (Uchiyama et al., 2013) and lysosomal capacity impaired (Shim et al., 2016). This feature is common to most protein misfolding neurodegenerative disorders (Koh et al., 2019), and now, through this new evidence, it is directly linked to the interaction of these misfolded proteins with PrPc.
An indication, perhaps, that the true role of PrPc is involved in the quality control and sorting of proteins for reuse versus degradation based on their correctly folded or misfolded state?
References:
Peggion C, Stella R, Chemello F, Massimino ML, Arrigoni G, Cagnin S, Biancotto G, Franchin C, Sorgato MC, Bertoli A. The Prion Protein Regulates Synaptic Transmission by Controlling the Expression of Proteins Key to Synaptic Vesicle Recycling and Exocytosis. Mol Neurobiol. 2018 Aug 20; PubMed.
Heisler FF, Pechmann Y, Wieser I, Altmeppen HC, Veenendaal L, Muhia M, Schweizer M, Glatzel M, Krasemann S, Kneussel M. Muskelin Coordinates PrPC Lysosome versus Exosome Targeting and Impacts Prion Disease Progression. Neuron. 2018 Sep 19;99(6):1155-1169.e9. Epub 2018 Aug 30 PubMed.
Uchiyama K, Miyata H, Sakaguchi S. Disturbed vesicular trafficking of membrane proteins in prion disease. Prion. 2013 Dec 11;7(6) PubMed.
Shim SY, Karri S, Law S, Schatzl HM, Gilch S. Prion infection impairs lysosomal degradation capacity by interfering with rab7 membrane attachment in neuronal cells. Sci Rep. 2016 Feb 11;6:21658. PubMed.
Koh JY, Kim HN, Hwang JJ, Kim YH, Park SE. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: possible therapeutic roles of cAMP and zinc. Mol Brain. 2019 Mar 12;12(1):18. PubMed.
University of Edinburgh
This interesting study overlooks that PrP, like Alzheimer Aβ (Soscia et al., 2010; Moir et al., 2018), and probably also α-synuclein (Park et al., 2016), is an antimicrobial protein (AMP)—as pointed out by Artur Schmidtchen and colleagues in Sweden (Pasupuleti et al. ,2009, overviewed in Lathe and Darlix, 2017; Lathe and Darlix, 2020)—that directly interacts with other AMPs including Aβ, the archetypical AMP LL-37, and probably also Parkinson's disease α-synuclein. The protection afforded by AMPs is a delicate balance between pathogen-specific toxicity and toxicity to host cells if overexpressed (reviewed by Rob Moir and colleagues in Moir et al., 2018). We again risk confusing defense with causation, as has been the case for Aβ. Perhaps let's not blame the defenders of the brain for causing the unrest quite yet. That said, many infectious agents (from HIV to hepatitis C to cytomegalovirus) exploit AMPs; herpes simplex virus has developed a mechanism to sidestep PrP-mediated inhibition, and instead uses PrP to boost its own replication (Lathe and Darlix, 2017). However, the evidence that PrP is involved at all, as Walsh et al. report (key article), surely adds further fuel to the emerging consensus that pathogens are involved in the pathoetiology of AD.
References:
Lathe R, Darlix JL. Prion Protein PRNP: A New Player in Innate Immunity? The Aβ Connection. J Alzheimers Dis Rep. 2017 Dec 16;1(1):263-275. PubMed.
Lathe R, Darlix JL. Prion protein PrP nucleic acid binding and mobilization implicates retroelements as the replicative component of transmissible spongiform encephalopathy. Arch Virol. 2020 Mar;165(3):535-556. Epub 2020 Feb 5 PubMed.
Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018 Dec;14(12):1602-1614. Epub 2018 Oct 9 PubMed.
Park SC, Moon JC, Shin SY, Son H, Jung YJ, Kim NH, Kim YM, Jang MK, Lee JR. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. 2016 Sep 16;478(2):924-8. Epub 2016 Aug 9 PubMed.
Pasupuleti M, Roupe M, Rydengård V, Surewicz K, Surewicz WK, Chalupka A, Malmsten M, Sörensen OE, Schmidtchen A. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. 2009;4(10):e7358. PubMed.
Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010 Mar 3;5(3):e9505. PubMed.
Make a Comment
To make a comment you must login or register.